6 Dagens regulering og forvaltning av genmodifiserte organismer og avledete produkter
6.1 Innledning
Norge er gjennom EØS-avtalen tett knyttet til EUs GMO-regelverk, som består av fire hovedrettsakter; Europaparlaments- og rådsdirektiv 2001/18/EF av 12. mars 2001 om utsetting i miljøet av genmodifiserte organismer og oppheving av rådsdirektiv 90/220/EØF (utsettingsdirektivet), Europaparlaments- og rådsdirektiv 2009/41/EF av 6. mai 2009 om innesluttet bruk av genmodifiserte mikroorganismer (innesluttet bruk-direktivet), Europaparlaments- og rådsforordning (EF) nr. 1829/2003 av 22. september 2003 om genmodifisert mat og fôr (GM mat- og fôrforordningen) og forordning (EF) nr. 1830/2003 av 22. september 2003 om sporbarhet og merking av genmodifiserte organismer og om sporbarhet av mat og fôr fremstilt på grunnlag av genmodifiserte organismer, og endring av direktiv 2001/18/EF (sporbarhets- og merkeforordningen). Utsettingsdirektivet og innesluttet bruk-direktivet er innlemmet i EØS-avtalen og tatt inn i norsk rett gjennom genteknologiloven, mens de to forordningene ikke er gjennomført i norsk rett per i dag. Genteknologiloven bygger i stor grad på bestemmelser i de to direktivene, mens hovedelementene i EUs regelverk om genmodifisert mat og fôr er inkludert i nasjonalt regelverk under matloven som regulerer prosesserte (det vil si ikke-spiredyktige) produkter laget fra GMO.
6.1.1 GMO-forvaltningen i Norge – sektoransvar
I Norge er forvaltningen av GMO-området delt mellom flere departementer og underliggende etater.
Klima- og miljødepartementet har hovedansvaret for genteknologiloven og for forvaltningen av levende GMO. Helse- og omsorgsdepartementet har ansvar for innesluttet bruk av GMO og kloning av dyr etter genteknologiloven. Kloning av dyr omfattes ikke av utvalgets mandat. Helse- og omsorgsdepartementet er også ansvarlig myndighet for legemiddelloven. De tre matdepartementene Helse- og omsorgs-, Landbruks- og mat- og Nærings- og fiskeridepartementet er sammen ansvarlige for matloven og regelverket om genmodifisert mat og fôr med hjemmel i matloven.
Norge er gjennom EØS-avtalen tilknyttet EUs godkjenningsordning for søknader om godkjenning av levende GMO mottatt etter utsettingsdirektivet, og Klima- og miljødepartementet er kompetent myndighet for direktivet og har vedtaksmyndighet for GMO til utsetting, inkludert omsetning (med de unntak som fremgår under punkt 6.1.2 nedenfor). EUs regelverk for genmodifisert mat og fôr er foreløpig ikke innlemmet i EØS-avtalen, men regelverket er hjemlet i EUs matlov som er innlemmet i norsk rett med Mattilsynet som kompetent myndighet.
6.1.2 Direktoratsnivået
På direktoratsnivå har følgende etater vedtaksmyndighet:
Helsedirektoratet for søknader om innesluttet bruk av levende GMO etter genteknologiloven
Miljødirektoratet for søknader om forsøksutsetting, samt utsetting i veksthus, akvakulturinnretning, dyrestaller og lignende som ikke er godkjent for innesluttet bruk, av levende GMO etter genteknologiloven
Legemiddelverket for søknader om godkjenning av GMO i kliniske utprøvinger etter legemiddelloven og, som en ordning frem til forslagene fra dette GMO-utvalget er lagt frem og er nærmere vurdert1, for søknader om klinisk utprøving av GMO-legemidler til mennesker og dyr etter genteknologiloven.
Mattilsynet for søknader om godkjenning av mat og fôr fra GMO etter matloven, for søknader om godkjenning av GMO-plantesorter etter matloven, for søknader om bruk av forsøksdyr, herunder også pre-kliniske forsøk med GMO-legemidler til dyr og mennesker etter dyrevelferdsloven, dyrehelsepersonelloven og matloven, og for godkjenning av virksomheter som driver med forsøksdyr etter dyrehelsepersonelloven.
Ansvarsområdene til de enkelte direktorater og tilsyn:
Miljødirektoratet
Ivaretar miljøhensyn ved utsetting av GMO i miljøet som ansvarlig for miljørisikohåndtering av GMO, herunder oppdrag om miljørisikovurdering til Vitenskapskomiteen for mat og miljø (heretter VKM), og som tilsynsmyndighet for GMO som ikke er til mat og fôr
Har vedtaksmyndighet for utsetting av GMO til andre formål enn omsetning etter genteknologiloven. Dette omfatter også utsetting i forskningsøyemed (feltforsøk med mer), med unntak av utsetting av GMO-legemidler til utprøving i kliniske studier
Ansvarlig for å uttale seg om miljørisiko til Legemiddelverket i forbindelse med søknader om klinisk utprøving av GMO-legemidler
Vurdering/vedtak om import, transport, eksport av GMO
Koordinerer nasjonal søknadsprosedyre for søknader om omsetning mottatt etter utsettingsdirektivet i EU, og utarbeider tilrådning om disse til Klima- og miljødepartementet
Har delegasjonsansvar for møter i EUs faste komité under utsettingsdirektivet
Gir innspill til Det europeiske legemiddelbyrået (EMA) om eventuell miljørisiko og miljørisikohåndteringstiltak for GMO-legemidler det søkes omsetningstillatelse for
Deltar i EUs sentraliserte prosedyre for søknader til EU om godkjenning av genmodifisert mat og fôr under GM mat- og fôrforordningen
Deltar i EU/EØS regelverks- og forvaltningsarbeid på oppdrag fra Klima- og miljødepartementet
Deltar i internasjonalt arbeid under Cartagena-protokollen, og er nasjonalt kontaktpunkt for «Biosafety Clearing House»2 (åpen database for utveksling av informasjon om GMO)
Mattilsynet
Ivaretar trygghet for helse ved GMO og prosesserte mat- og fôrprodukter fra GMO som ansvarlig for helserisikohåndtering av GMO til omsetning etter matloven og genteknologiloven, herunder oppdrag om helserisikovurdering til VKM
Ivaretar trygghet for agronomisk miljørisiko ved GMO-planter til dyrking, herunder sameksistens og med oppdrag til VKM
Har vedtaksmyndighet for genmodifisert mat og fôr fra GMO etter matloven, vedtaksmyndighet for godkjenning av genmodifiserte plantesorter for opptak på norsk offisiell sortsliste og er ansvarlig myndighet for sertifisering av genmodifiserte såvarer produsert i Norge
Er tilsynsmyndighet for levende og prosessert GMO til mat og fôr og gjennomfører den offentlige kontrollen med hjemmel i henholdsvis genteknologiloven og matloven
Har delegasjonsansvar for møter i EUs faste komité for genmodifisert mat og fôr
Deltar i EUs sentraliserte prosedyre for søknader til EU om godkjenning av genmodifisert mat og fôr under GM mat- og fôrforordningen
Deltar i EU/EØS regelverks- og forvaltningsarbeid på oppdrag fra matdepartementene
Er nasjonalt kontaktpunkt for Codex Alimentarius3
Er nasjonalt kontaktpunkt for FAO GM Foods Platform
Har delegasjonsansvar for møter i «The International Union for the Protection of New Varieties of Plants» (UPOV)4
Legemiddelverket
Vurderer nytte/risiko i søknader om klinisk utprøving av legemidler til mennesker og til dyr, og har vedtaksmyndighet etter legemiddelloven for disse søknadene
Er nasjonalt kontaktpunkt for vurdering og godkjenning av klinisk utprøving av legemidler etter legemiddelregelverket
Har siden 15. november 2021 hatt vedtaksmyndighet etter § 10 for GMO-legemidler til utprøving i kliniske studier til mennesker og dyr etter genteknologiloven
Deltar i EUs sentraliserte prosedyre for søknader om markedsføring/omsetning av legemidler og har vedtaksmyndighet for søknader om markedsføring av alle legemidler inkludert GMO-legemidler til mennesker og dyr. Dette inkluderer GMO-utredning.
Har ansvar for vurderingen av risiko/nytte-forholdet ved søknad om markedsføringstillatelse. Her inngår det en vurdering av miljørisiko etter legemiddelregelverket.
Deltar i EU/EØS regelverks- og forvaltningsarbeid på oppdrag fra Helse- og omsorgsdepartementet
Deltar i alle vitenskapelige komiteer administrert av EMA (Det europeiske legemiddelbyrået)
Har ansvar for forvaltningsoppgaver som gjelder forsyningskjeden for medisinske produkter
Har forvaltningsansvar for legemiddeløkonomi, herunder fastsette pris på reseptbelagte legemidler til mennesker, vurdere om legemidler er kostnadseffektive, og innkreving av avgifter knyttet til omsetning av legemidler, gi apotek driftsstøtte og fraktrefusjon og utarbeide apotekstatistikk
Har forvaltningsansvar for Medisinsk utstyr
Har ansvar for tilsyn og overvåkning med aktørene, herunder produsenter, blodbanker, importører, grossister og apotek, og de som er ansvarlige for kliniske legemiddelutprøvinger.
Helsedirektoratet
Ivaretar trygghet for helse og miljø fra GMO til innesluttet bruk
Er vedtaks- og tilsynsmyndighet for innesluttet bruk av GMO etter genteknologiloven. Denne myndigheten omfatter også klinisk utprøving av legemidler til mennesker og dyr
Har ansvar for oppfølging av EUs innesluttet bruk-direktiv
6.1.3 Andre instanser
Av andre instanser med definerte roller, har VKM en viktig rolle som risikovurderingsorganet for myndighetene etter genteknologiloven og matloven. VKM utfører helse- og miljørisikovurdering av GMO som søkes godkjent i Norge eller i EU på oppdrag fra Mattilsynet, Miljødirektoratet og Legemiddelverket.
Bioteknologirådet har en lovfestet rolle etter genteknologiloven for vurdering av kriteriene bærekraft, samfunnsnytte og etikk og kan på eget initiativ, eller på begjæring, levere råd om GMO til myndighetene. Rådets rolle i GMO-søknader fremmet under utsettingsdirektivet er beskrevet i de fastsatte saksbehandlingsrutinene5. Se mer informasjon om Bioteknologirådet når det gjelder GMO-legemidler i kapittel 6.3.2.2.
Regionale komiteer for medisinsk og helsefaglig forskningsetikk (REK), og senere Komiteene for klinisk utprøving av legemidler og medisinsk utstyr (REK KULMU) har et nasjonalt mandat der komiteene skal foreta en forskningsetisk vurdering av blant annet søknader som omhandler klinisk utprøving av legemidler. REK hadde dette mandatet tidligere, og REK KULMU ble etablert høsten 2020 for å imøtekomme Norges forpliktelser til oppfølging av blant annet felles regelverk i EU/EØS for klinisk utprøving av legemidler til mennesker.
6.1.4 Nærmere oversikt over innholdet i kapittel 6
I det følgende beskrives genteknologiloven (kapittel 6.2), regelverk som gjelder GMO-legemidler (både legemiddel- og genteknologiloven; kapittel 6.3), matlovsregelverk (kapittel 6.4), den offentlige kontrollen i Norge med hensyn til genmodifisering i mat, fôr og såvarer, samt deteksjonsmetoder og navnsetting av GMO med videre (kapittel 6.5.) Tilgrensende matlovsregelverk omtales i kapittel 6.6. Føre-var prinsippet og bruken av prinsippet i henhold til mat- og miljøområdet omtales spesielt i kapittel 6.7. Videre beskrives saksgangen i Norge for behandlingen av nasjonale søknader og søknader som sendes til EU (kapittel 6.8). Avslutningsvis beskrives koblingspunktene mellom GMO mat- og miljøregelverket i henholdsvis EU og Norge (kapittel 6.9 og 6.10).
De enkelte underkapitlene kan leses uavhengig av hverandre.
6.2 Genteknologiloven
I Norge reguleres framstilling og bruk av levende genmodifiserte organismer (GMO) av genteknologiloven. Klima- og miljødepartementet har det overordnede ansvaret for loven og for å behandle søknader om utsetting av levende GMO. Genmodifiserte prosesserte/bearbeidede produkter (det vil si antatt ikke-reproduserbare produkter) fra levende GMO, brukt til mat og fôr, reguleres under matloven. Genmodifiserte, prosesserte produkter som ikke kan brukes til mat eller fôr, er ikke regulert i Norge eller i EU, for eksempel bomullsfibre og klær laget fra GMO-bomull.
Genteknologiloven ble i sin tid utformet for å sikre en forsvarlig utvikling av genteknologi, hvor teknologien skulle brukes for samfunnets beste, i pakt med samfunnets verdier, og med de nødvendige begrensninger av hensyn til helse og miljø. Dette kan anses som reflektert i lovens oppbygning rundt føre-var-prinsippet, krav om ingen helse- og miljøskadevirkninger, og krav til vektlegging av etisk forsvarlighet, samfunnsnytteverdi og innvirkning på bærekraftig utvikling. Godkjenningsprosedyrene etter loven sikrer også medvirkning gjennom allmenn høring av søknader om utsetting, og valgfriheten til forhandlere og forbrukere gjennom krav om informasjon og merking av godkjente produkter som inneholder GMO.
Genteknologiloven inneholder også bestemmelser om kloning. Disse omtales ikke i kapittelet her da utvalgets mandat ikke omfatter kloning.
Regelverk og praksis som særlig gjelder GMO-legemidler omtales primært i kapittel 6.3 nedenfor. Omtalen omfatter informasjon om både legemiddelloven og genteknologiloven. Klinisk utprøving omtales i punkt 6.3.1.1. og 6.3.1.2, og markedsføring i punkt 6.3.1.3. Merk også at kapittel 12 er viet til GMO-legemidler.
6.2.1 Lovens formål, virkeområde, definisjoner m.m.
Lovens formål fremgår av § 1 og lyder:
Denne loven har til formål å sikre at framstilling og bruk av genmodifiserte organismer og framstilling av klonede dyr skjer på en etisk og samfunnsmessig forsvarlig måte, i samsvar med prinsippet om bærekraftig utvikling og uten helse- og miljømessige skadevirkninger.
Genteknologilovens formål er både å unngå helse- og miljømessige skadevirkninger, men også å sikre andre hensyn gjennom en forsvarlig utvikling av teknologien, det vil si etiske og samfunnsmessige hensyn og hensynet til en bærekraftig utvikling. Genteknologiloven skiller seg ved dette fra blant annet EØS-avtalens bestemmelser om framstilling og bruk av genmodifiserte organismer, som først og fremst regulerer risikomessige aspekter.
Begrepet uten helse- og miljømessige skadevirkninger må ifølge forarbeidene ikke oppfattes helt absolutt, jamfør Ot.prp. nr. 8 (1992–93) side 67.
Lovens saklige virkeområde fremgår av § 2. Her heter det i første ledd blant annet at loven gjelder framstilling og bruk av genmodifiserte organismer (…).
Det fremgår av annet ledd at:
Med mindre genmodifiserte organismer brukes som foreldreorganismer, gjelder loven ikke for framstilling ved hjelp av celleteknologi av
a) genmodifiserte planteceller når resultatet også kan oppnås ved tradisjonelle foredlingsmetoder, og
b) dyreceller i kultur der cellematerialet hentes fra ulike individer innen samme art og hvor cellene kunne oppstått ved naturlig formering,
samt bruk av slike plante- eller dyreceller.
Definisjonen av genteknologilovens virkeområde sammenholdt med definisjonen av begrepet genmodifiserte organismer i § 4 bokstav b, innebærer at genteknologiloven ikke omfatter genmodifisering av mennesker. Det fremgår av Ot.prp. nr. 8 (1992–93) om genteknologiloven side 69 at dette ikke gjelder humane celler i kultur. Det heter her: Definisjonen av organismer omfatter ikke mennesker på noe trinn i utviklingen fra befruktet egg til fullt utviklet individ, men den omfatter humane celler i kultur. Det er enighet også i EU om at humane genmodifiserte celler omfattes av GMO-regelverket, og således er tolkningen av regelverket på dette området i Norge harmonisert med EU.
Arbeid med arvematerialet utenfor en levende celle eller virus som ikke er et ledd i framstillingsprosessen av en organisme, faller også utenfor genteknologilovens virkeområde, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 70.
Forarbeidene presiserer videre at genteknologiloven kun omfatter framstilling og bruk av levende organismer (kapittel 9 side 69).
Framstilling av produkter ved hjelp av genmodifiserte organismer faller inn under genteknologiloven, siden dette må anses som bruk etter § 2 første ledd første punktum. Selve sluttproduktet faller derimot utenfor, med mindre dette selv inneholder levende genmodifiserte organismer, jamfør ordlyden i § 2 første ledd tredje punktum og Ot.prp. nr. 8 (1992–93) kapittel 9 side 69.
Paragraf 3 omhandler lovens stedlige virkeområde. Loven gjelder i riket, herunder Svalbard og Jan Mayen. Videre gjelder loven for de norske biland i Antarktis, innenfor Norges økonomiske sone og på den norske del av kontinentalsokkelen.
Paragraf 4 inneholder definisjoner, blant annet følgende definisjoner i bokstav a) til d):
a) mikroorganismer: enhver cellulær eller ikke-cellulær mikrobiologisk enhet som er i stand til å formere seg eller til å overføre genetisk materiale
b) genmodifiserte organismer: mikroorganismer, planter og dyr hvor den genetiske sammensetning er endret ved bruk av gen- eller celleteknologi
c) genteknologi: teknikker som innebærer at arvestoff isoleres, karakteriseres, modifiseres og innsettes i levende celler eller virus
d) celleteknologi: teknikker for framstilling av levende celler med nye kombinasjoner av genetisk materiale ved fusjon av to eller flere celler.
Definisjonen av mikroorganismer omfatter ifølge Ot.prp. nr. 8 (1992–93) virus, bakterier, encellede planter og dyr, plante- og dyreceller (herunder humane celler) i kultur og mikroskopiske gjær- og muggsopper (kapittel 9 side 70). Såkalt nakent genmateriale eller plasmider faller utenfor definisjonen.
Definisjonene i loven innebærer at en genmodifisert organisme er enhver organisme (foruten mennesker) som har fått endret sin genetiske sammensetning ved hjelp av gen- eller celleteknologi.
Definisjonen av genteknologi slår fast at dette er teknikker hvor arvestoff isoleres, karakteriseres, modifiseres og innsettes i levende celler eller virus. Forarbeidene til loven spesifiserer videre at modifisering av arvestoff også omfatter at en del av arvestoffet fjernes.
Forarbeidene fremhever videre at definisjonen av GMO etter loven utelukker organismer hvor det genetiske materiale er blitt endret på en måte som skjer naturlig ved formering og/eller naturlig rekombinasjon, ved tradisjonelle foredlingsmetoder, eller ved bruk av mutagener som kjemikalier eller stråling. Organismer fremkommet ved disse metodene omfattes derfor ikke av loven (jamfør Ot.prp. nr. 8 (1992–1993), side 69).
Ved genteknologiske metoder blir en eller flere egenskaper overført fra en organisme til en annen, eller en eller flere egenskaper i en organisme blir eliminert. Dette i motsetning til celleteknologien, der den nye organismen får alle genetiske egenskaper fra begge utgangsorganismer. Resultatet betegnes i begge tilfeller som en genmodifisert organisme etter genteknologiloven. I Ot.prp. nr. 8 (1992–93) blir det regnet opp hvilke konkrete teknikker som i hvert fall må anses som gen- eller celleteknologi etter genteknologiloven (side 71).
Utsettingsdirektivet har en noe annen GMO-definisjon enn genteknologiloven. I direktivet er genmodifisert organisme definert som:
en organisme, unntatt mennesker, der genmaterialet er endret på en måte som ikke forekommer ved naturlig formering og/eller naturlig rekombinasjon.
I tillegg er det i vedlegg til direktivet gitt en liste av teknikker som alltid gir en genmodifisert organisme til godkjenning etter direktivet (vedlegg IA, del 1). Disse er:
teknikker med rekombinasjon av nukleinsyre, som omfatter dannelse av nye kombinasjoner av genmateriale ved at nukleinsyremolekyler produsert på en hvilken som helst måte utenfor en organisme, settes inn i et virus, bakterieplasmid eller annet vektorsystem, samt at de innføres i en vertsorganisme som de ikke naturlig forekommer i, men der de er i stand til å fortsette å formere seg,
teknikker som innebærer direkte innføring i en organisme av arvestoffer som er tilberedt utenfor organismen, herunder mikroinjeksjon, makroinjeksjon og mikroinnkapsling,
cellefusjonsteknikker (herunder protoplastfusjon) eller hybridiseringsteknikker der levende celler med nye kombinasjoner av genetiske arvestoffer, dannes ved fusjon av to eller flere celler ved hjelp av metoder som ikke forekommer naturlig.
Det gis også en liste med teknikker som ikke gir en genmodifisert organisme etter definisjonen i direktivet:
befruktning in vitro,
naturlige prosesser som konjugasjon, transduksjon, transformasjon,
polyploid induksjon.
I tillegg gis det et unntak fra godkjenningsplikten etter direktivet for teknikker nevnt i vedlegg I B:
Teknikker/metoder for genmodifisering som gir opphav til organismer som skal utelukkes fra direktivets virkeområde, forutsatt at de ikke innebærer bruk av rekombinasjon av nukleinsyremolekyler eller genmodifiserte organismer unntatt dem som er produsert med en eller flere av teknikkene/metodene oppført nedenfor, er:
1. mutagenese,
2. cellefusjon (herunder protoplastfusjon) av planteceller fra organismer som kan utveksle genmateriale ved tradisjonelle foredlingsmetoder.
Departementet tok ved utformingen av genteknologiloven stilling til definisjonsspørsmålet, og valgte den lovtekniske løsningen å utelukke metodene nevnt i utsettingsdirektivets vedlegg I B, direkte gjennom GMO-definisjonen til loven.
Definisjonen av genmodifiserte organismer er et aktuelt tema både i Norge og EU. For noen nye teknikker ble det klargjort om de faller innenfor eller utenfor definisjonen med EU-domstolens dom i 2018, C-528/16. Domstolen slo da fast at organismer som modifiseres ved hjelp av nye mutageneseteknikker, og gir målrettede endringer i en organismes egne gener, blir en genmodifisert organisme etter utsettingsdirektivets definisjon om genmodifiserte organismer. Det andre spørsmålet var om slike genmodifiserte organismer skulle unntas godkjenning etter utsettingsdirektivet, i likhet med konvensjonell mutagenese ved ioniserende stråling og kjemikalier. EU domstolen konkluderte her med at nye mutageneseteknikker ikke kunne unntas godkjenning på samme grunnlag som de konvensjonelle teknikkene. I dommen avsnitt 51 står det i dansk oversettelse:
Under disse omstændigheder kan artikel 3, stk. 1, i direktiv 2001/18, (unntaksbestemmelsen, vår anmerking) sammenholdt med nr. 1) i bilag I B til dette direktiv, ikke fortolkes således, at den fra dette direktivs anvendelsesområde udelukker organismer fremstillet ved hjælp af nye mutageneseteknikker/-metoder, der er opstået eller hovedsageligt er blevet udviklet siden vedtagelsen af det nævnte direktiv. En sådan fortolkning ville nemlig føre til, at EU-lovgivers hensigt, der er afspejlet i 17. betragtning til dette direktiv om fra direktivets anvendelsesområde kun at udelukke organismer fremstillet ved hjælp af teknikker/metoder, som traditionelt er blevet brugt i en række anvendelser, og som gennem lang tid har vist sig sikre, blev tilsidesat.
Som tidligere nevnt må Norge ha et regelverk som innholdsmessig er i overensstemmelse med direktivet.
Hverken utsettingsdirektivets eller genteknologilovens GMO-definisjon stiller noe krav om at den genetiske endringen skal være arvelig eller permanent.
I 2017 vurderte Europakommisjonen i konsultasjon med medlemsstatene om fisk vaksinert med DNA-vaksinen Clynav, skulle defineres som GMO. Clynav er den første DNA-vaksinen til dyr i Europa, og brukes mot pankreassykdom hos laks.6 Clynav består av et DNA-plasmid som koder for proteiner fra salmanoid alfavirus som gir immunitet mot viruset. Se mer detaljer i boks 8.9. Selve Clynav-vaksinen inneholder ikke GMO, og faller utenfor genteknologiloven. I sin beslutning 27. juni 2017 konkluderte Kommisjonen med at fisk vaksinert med Clynav, ikke skal defineres som GMO. Det ble samtidig fremhevet at konklusjonen ikke gjelder for alle DNA-vaksiner, men at hver vaksine vil måtte vurderes individuelt fra sak til sak. EØS-avtalens mål om et velfungerende indre marked forutsetter mest mulig enhetlig gjennomføring av avtalens regler i alle EØS-landene. På denne bakgrunn besluttet Klima- og miljødepartementet i 2017 at det også skulle være norske myndigheters forståelse at fisk vaksinert med Clynav ikke skal defineres som GMO, og ikke omfattes av genteknologiloven.
Dette er i utgangspunktet ikke i samsvar med definisjonen av genmodifiserte organismer i genteknologiloven § 4 b), fordi den genetiske sammensetningen hos DNA-vaksinerte fisk blir endret ved bruk av gen- eller celleteknologi, selv om denne endringen ikke er varig.
6.2.2 Innesluttet bruk – definisjon og sikkerhetstiltak
Genteknologiloven regulerer som nevnt både utsetting og innesluttet bruk. Hva som skal anses som innesluttet bruk, fremgår av § 5, som slår fast at med innesluttet bruk menes enhver arbeidsoperasjon hvor genmodifiserte organismer blir framstilt, dyrket, lagret, destruert eller brukt på annen måte, i et lukket system hvor det anvendes fysiske inneslutningstiltak, eventuelt i kombinasjon med andre særskilte inneslutningstiltak, for å begrense organismenes kontakt med mennesker og miljø slik at disse sikres et høyt nivå av sikkerhet.
Definisjonen av innesluttet bruk følger av rådsdirektiv 98/81/EF (senere erstattet av innesluttet bruk-direktivet uten at definisjonen ble endret).
Det sentrale i definisjonen av innesluttet bruk, er at virksomheten skal foregå i et lukket system, for å hindre at organismene eller deres arvemateriale sprer seg, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 71–72, og Ot.prp. nr. 60 (2000–2001) kapittel 2.2. Kravet om et lukket system ble opprettholdt ved lovendringen i 2001, selv om begrepet ikke lenger brukes i definisjonen i innesluttet bruk-direktivet. For å oppnå dette, kreves i det minste fysiske barrierer, det vil si den fysiske utforming av laboratoriet, produksjonsutstyr med videre.
Mens Klima- og miljødepartementet har det overordnede ansvaret for loven, og for utsetting, er ansvaret for innesluttet bruk lagt til Helse- og omsorgsdepartementet.
Paragraf 6 omhandler sikkerhetstiltak ved innesluttet bruk. Det fremgår av første ledd at innesluttet bruk skal skje i laboratorier og anlegg som er godkjent av Kongen, og i samsvar med god mikrobiologisk praksis. Brukeren skal sørge for de nødvendige sikkerhetstiltak for å hindre helse- og miljømessige skadevirkninger, herunder tiltak for å begrense skadevirkninger ved utilsiktet utslipp av genmodifiserte organismer. Det skal føres protokoll over all innesluttet bruk av genmodifiserte organismer.
God mikrobiologisk praksis må ifølge forarbeidene oppfattes som en rettslig standard for håndtering og bruk av mikrobiologiske forsøk. De konkrete krav som må stilles vil variere fra sak til sak, og ha sitt grunnlag i uskrevne eller eventuelt skrevne retningslinjer i de miljøene som arbeider med den aktuelle typen mikrobiologisk virksomhet. Sammen med § 6 første ledd annet punktum oppstiller kravet om god mikrobiologisk praksis en aktsomhetsplikt for den enkelte forsker eller virksomhet. Se Ot.prp. nr. 8 (1992–93) kapittel 9 side 72-73.
Det følger av annet ledd at laboratorier og andre anlegg for innesluttet bruk av genmodifiserte organismer skal være godkjent av Kongen.
Tredje og fjerde ledd slår fast at Kongen kan gi forskrift om sikkerhetstiltak ved innesluttet bruk og om det nærmere innhold i plikten til å føre protokoll. Kongen kan ved forskrift også gjøre unntak fra regler i paragraf 6 for nærmere bestemte former for undervisningsvirksomhet. Slike bestemmelser er gitt i forskriftene 21. desember 2001 nr. 1600 (forskrift om innesluttet bruk av genmodifiserte mikroorganismer), 21. desember 2001 nr. 1601 (forskrift om bestemte former for undervisningsvirksomhet som innebærer innesluttet bruk av genmodifiserte mikroorganismer), 21. desember 2001 nr. 1602 (forskrift om innesluttet bruk av genmodifiserte dyr) og 21. desember 2001 nr. 1603 (forskrift om innesluttet bruk av genmodifiserte planter).
6.2.3 Innesluttet bruk – meldeplikt eller godkjenning
Det følger av § 7 første ledd at innesluttet bruk av genmodifiserte organismer skal meldes eller godkjennes i samsvar med forskrift gitt av Kongen. Det kan i forskriften gjøres unntak for nærmere bestemte former for undervisningsvirksomhet.
Utgangspunktet er at all innesluttet bruk enten skal være meldepliktig eller avhengig av særskilt tillatelse. Slik melding eller tillatelse kommer i tillegg til kravet om laboratoriegodkjenning etter § 6 annet ledd. Om virksomhet er melde- eller godkjenningspliktig er enten avhengig av risikoen ved virksomheten, eller av særskilte etiske og samfunnsmessige vurderinger. Inndeling i melde- eller godkjenningspliktig virksomhet skjer i utgangspunktet ved forskrift, jamfør § 7 første ledd første punktum. Bestemmelser er gitt i forskrift 21. desember 2001 nr. 1600 om innesluttet bruk av genmodifiserte mikroorganismer, forskrift 21. desember 2001 nr. 1601 om bestemte former for undervisningsvirksomhet som innebærer innesluttet bruk av genmodifiserte mikroorganismer, forskrift 21. desember 2001 nr. 1602 om innesluttet bruk av genmodifiserte dyr og forskrift 21. desember 2001 nr. 1603 om innesluttet bruk av genmodifiserte planter.
Lovgiveren har i § 7 annet ledd regnet opp en del tilfeller der det av etiske og samfunnsmessige grunner kreves særskilt godkjenning uansett risikomessige vurderinger. For begrunnelsen av disse særreglene, se Ot.prp. nr. 8 (1992–93) kapittel 4.2 side 34 og kapittel 4.14 side 46, St.meld. nr. 8 (1990–91) og Innst. S. nr. 155 (1990-91).
Etter annet ledd kreves det godkjenning for følgende former for innesluttet bruk:
a) genetisk modifisering av virveldyr som innebærer arvelige endringer med mindre det gjelder forsøk som er godkjent etter dyrevelferdsloven § 13
b) overføring av humant genmateriale til dyr, planter eller mikroorganismer, som ikke skjer i forsknings- eller forsøkssammenheng for å kartlegge arvestoffets oppbygging, egenskaper og funksjoner
c) framstilling og bruk av genmodifiserte organismer for omsetning eller annen utnytting i næring.
Med virveldyr menes pattedyr, fugler, fisk, amfibier og krypdyr, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 75. Mennesker omfattes ikke av genteknologiloven, med unntak av humane genmodifiserte celler og nevnte bruk av humant genmateriale.
Humant genmateriale er ifølge Ot.prp. nr. 8 (1992–93) genmateriale som koder for prosesser og egenskaper på en måte som foregår i mennesker, og som har sin opprinnelse fra menneskeceller.
Paragraf 7 annet ledd bokstav c gjelder den industrielle framstillingsprosessen som resulterer i genmodifiserte organismer, eller som skjer ved hjelp av genmodifiserte organismer. Selve omsetningen regnes imidlertid som utsetting, og reguleres etter lovens kapittel 3.
Kongen kan etter tredje ledd gi forskrift om at produksjon som nevnt i annet ledd bokstav c for bestemte typer eller mengder av genmodifiserte organismer i stedet skal være meldepliktig.
Fjerde ledd slår fast at reglene om meldeplikt og krav om godkjenning etter § 7 gjelder ikke for framstilling og bruk av dyrehybridceller for produksjon av monoklonale antistoffer eller for isolering av kromosomer og kromosomfragmenter. Dyrehybridceller er en sammensmelting av to dyreceller fra forskjellige arter. De cellene som brukes for framstilling av monoklonale (det vil si ensartede eller identiske) antistoffer, er framkommet ved sammensmelting av en kreftcelle og en celle som produserer et bestemt antistoff, jamfør Ot.prp. nr. 8 (1992–93) kapittel 4.5 side 36–37.
Paragraf 8 slår fast at Kongen kan kreve at den som søker om godkjenning av innesluttet bruk, skal sørge for en konsekvensutredning for å klarlegge konsekvensene av utilsiktede utslipp av genmodifiserte organismer.
Konsekvensutredning kan ikke kreves ved innesluttet bruk som kun er meldepliktig. Blir det først krevd konsekvensutredning ved innesluttet bruk, kreves imidlertid i prinsippet de samme opplysninger som ved en konsekvensutredning i forbindelse med utsettingssøknad, jamfør forskrift 16. desember 2005 nr.1495 om konsekvensutredning etter genteknologiloven.
6.2.4 Utsetting
Hva som skal anses som utsetting i loven er definert i § 9 første ledd. Med utsetting forstås enhver framstilling og bruk av genmodifiserte organismer som ikke regnes som innesluttet bruk. Definisjonene av innesluttet bruk og utsetting er komplementære. Det vil si at det er snakk om utsetting når virksomheten ikke foregår i et lukket system med tilstrekkelige barrierer for å unngå spredning av organismene eller deres genmateriale, jamfør Ot.prp. nr. 8 (1992–93) kapittel 5.4 side 54 og kapittel 9 side 76.
De viktigste eksemplene på hva som er å forstå som en utsetting regnes opp i § 9 annet ledd.
Disse omfatter:
a) utsetting av genmodifiserte organismer i forskningsøyemed (feltforsøk)
b) utsetting av genmodifiserte organismer i næringsøyemed, til opprenskningsformål, o.l.
c) bruk av genmodifiserte organismer i veksthus, akvakulturinnretning, dyrestaller o.l., med mindre slike er godkjent for innesluttet bruk som en del av et godkjent laboratorium eller annet anlegg
d) rutineutslipp av genmodifiserte organismer fra innesluttet bruk
e) deponering av avfall som inneholder levende genmodifiserte organismer
f) omsetning av et produkt som består av eller som inneholder genmodifiserte organismer
g) import av genmodifiserte organismer
h) transport av genmodifiserte organismer
i) eksport.
Rutineutslipp i bokstavledd d) vil si bevisste utslipp fra innesluttet bruk, i motsetning til utilsiktede utslipp ved uhell, driftsforstyrrelser med mer, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 77.
6.2.5 Vurdering av helseeffekter og miljøpåvirkning
Paragraf 11 inneholder bestemmelser om konsekvensutredning. Det følger av første ledd at søknad om godkjenning av en utsetting skal inneholde konsekvensutredning for å klarlegge risikoen for helse- og miljømessige skadevirkninger og andre følger av utsettingen. Kongen kan i forskrift gi bestemmelser om blant annet utredningens innhold og om unntak fra utredningsplikten.
Andre følger sikter til de mer samfunnsmessige konsekvensene av utsettingen, både positive og negative, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 82.
Kongen kan etter § 11 annet ledd i tillegg til konsekvensutredningen kreve ytterligere opplysninger og undersøkelser før søknaden avgjøres.
Forskrift om konsekvensutredning etter genteknologiloven regulerer innholdet i og behandlingen av konsekvensutredningen som skal utarbeides ved godkjenningspliktig utsetting av genmodifiserte organismer.
Forskriften ble sist oppdatert i juni 2020, som følge av innlemmelse av direktiv (EU) 2018/350 som endrer utsettingsdirektivets vedlegg om prinsippene for miljørisikovurdering og informasjon for genmodifiserte, høyerestående planter.
Reglene om konsekvensutredninger fastsetter krav til myndighetene om saksbehandling og vedtak, og til søker om innhold i søknad og utforming av konsekvensutredningen.
Konsekvensutredningen skal i nødvendig utstrekning og så langt det er mulig gi en beskrivelse av det tiltak det søkes godkjenning for, og redegjøre for risikoen for og omfanget av de miljø- og folke- og dyrehelsemessige virkninger som kan oppstå på grunn av tiltaket og ved utilsiktet spredning fra tiltaket. Konsekvensutredningen skal også i nødvendig utstrekning og så langt det er mulig i samsvar med vedlegg 4 redegjøre for andre følger av tiltaket enn de miljø- og folke- og dyrehelsemessige virkninger, herunder:
1. tiltakets positive eller negative virkning for bærekraftig utvikling,
2. etiske aspekter som kan reises ved bruk av den genmodifiserte organisme og
3. eventuelle samfunnsmessige fordeler eller ulemper ved bruk av de genmodifiserte organismene.
Konsekvensutredningen skal sikre at søker tar disse i betraktning forut for gjennomføringen av tiltaket og at godkjenningsmyndigheten får et best mulig beslutningsgrunnlag når den skal ta stilling til om, og eventuelt på hvilke vilkår, godkjenning kan gis.
Informasjon som skal i søknaden fremgår av vedlegg 1 til forskriften, mens utforming av helse- og miljørisikovurderingen fremgår av vedlegg 2, og utforming av overvåkningsplan fremgår av vedlegg 3. Disse tre vedleggene samsvarer med innholdet i utsettingsdirektivet.
En sentral del av konsekvensutredningen er en helse- og miljørisikovurdering. Ifølge forskrift om konsekvensutredning etter genteknologiloven vedlegg 2 om prinsipper for miljørisikovurdering er formålet med en miljørisikovurdering av genmodifiserte organismer å identifisere og vurdere i hvert enkelt tilfelle den genmodifiserte organismens mulige skadevirkninger, enten direkte eller indirekte, umiddelbare eller over lang tid, på menneskers helse og på miljøet, som utsetting eller markedsføring av genmodifiserte organismer kan medføre. Miljørisikovurderingen bør utføres med sikte på å fastslå hvorvidt det er behov for risikostyring, og i så fall, hvilke metoder som vil være mest hensiktsmessige.
Miljørisikovurderingen gjøres i utgangspunktet av den medlemsstat som mottar søknaden. Deretter sendes søknaden på høring til øvrige EØS-land. For søknader om omsetning gjelder at i de tilfeller hvor ett eller flere EØS-land eller EU-kommisjonen har innsigelser, kommentarer eller spørsmål om søkers frembragte opplysninger om risiko for menneskers helse eller miljø, skal EU-kommisjonen be EUs risikovurderingsorgan «The European Food Safety Authority» (EFSA7; EUs mattrygghetsorgan) om en vitenskapelig vurdering av disse forhold. Til nå har alle søknader om omsetning i praksis blitt vurdert av EFSA. Miljørisikovurderingen gjøres etter utsettingsdirektivet, helserisikovurderingen etter forordning (EU) nr. 503/2013 og EFSAs egne retningslinjer for helse- og miljørisikovurderinger.
At genmodifiserte organismer ikke skal medføre helse- og miljøskade er et sentralt formål med loven. Vurdering av slik risiko skal skje på forhånd, og man skal unngå eventuelle skadevirkninger ved å sette inn relevante kontrolltiltak. I de tilfeller hvor det er en rimelig grad av tvil om risiko, skal føre-var-prinsippet legges til grunn for å unngå skadevirkninger for helse og miljø.
Forskrift om konsekvensutredning etter genteknologiloven inneholder generelle prinsipper som skal følges når miljørisikovurderingen foretas. Miljørisikovurdering er nærmere omtalt i kapittel 8. Prinsippene bygger på EUs utsettingsdirektiv og innebærer blant annet at:
Kjente egenskaper ved den genmodifiserte organismen, og ved bruken av den, som muligvis kan gi skadevirkninger, bør sammenlignes med egenskapene til de ikke-modifiserte organismene som den genmodifiserte organismen er avledet fra, og med bruken av disse under tilsvarende forhold. Denne metoden refereres til som komparativ analyse.
Miljørisikovurderingen bør foretas på en vitenskapelig forsvarlig og transparent måte og være basert på tilgjengelige vitenskapelige og tekniske data.
Det bør foretas en miljørisikovurdering i hvert enkelt tilfelle, da det kan være store ulikheter med hensyn til hvilke opplysninger som kreves, avhengig av hvilken type genmodifisert organisme det dreier seg om, hvilken bruk de er tiltenkt samt de mulige mottaksmiljøene, idet det blant annet tas hensyn til genmodifiserte organismer som allerede finnes i dette miljøet.
Dersom nye opplysninger om den genmodifiserte organismen og dens virkninger på menneskers helse og på miljøet blir tilgjengelige, kan det bli nødvendig å gjenta miljørisikovurdering, slik at
det kan fastslås om risikoen har endret seg,
det kan fastslås om det er behov for å endre risikostyringen tilsvarende.
I forskrift om konsekvensutredning etter genteknologiloven åpnes det for at konsekvensutredningen ikke trenger å være like omfattende i alle tilfeller, jamfør den innledende ordlyden i vedlegg 1.
6.2.6 Hensyn til bærekraft, samfunnsnytte og etikk
Etter genteknologiloven § 10 annet ledd kan utsetting av genmodifiserte organismer bare godkjennes når det ikke foreligger fare for miljø- og helsemessige skadevirkninger. Dessuten skal det ved avgjørelsen av en søknad legges vesentlig vekt på om utsettingen har samfunnsmessig nytteverdi og er egnet til å fremme en bærekraftig utvikling. Også etiske hensyn skal tillegges vekt, jamfør § 1 om lovens formål. Første punktum er en mer absolutt retningslinje for forvaltningens skjønnsutøvelse enn annet punktum, jamfør ordene kan bare godkjennes når det ikke foreligger.
Bærekraft, samfunnsnytte og etikk er særnorske kriterier, og veiledning til vurdering av disse for en GMO-søknad omsøkt til Norge er gitt i forskrift om konsekvensutredning etter genteknologiloven vedlegg 4.
Konsekvensutredningen skal i tillegg til helse og miljø redegjøre i nødvendig utstrekning om bruken av genmodifiserte organismer har samfunnsnytteverdi, er etisk forsvarlig og har negativ eller positiv virkning for bærekraftig utvikling. I kommentarene til § 10 annet ledd på side 81 i Ot.prp.nr. 8 (1992–1993) fremgår det at genmodifiserte organismers samfunnsnytte og bidrag til en bærekraftig utvikling både er selvstendige kriterier for vurdering av søknader etter loven og kriterier som kan bidra til en oppmyking av kravet om at utsetting av genmodifiserte organismer ikke skal ha helse- eller miljømessige skadevirkninger.
Forarbeidene inneholder en del veiledning med hensyn til hva en etisk og samfunnsmessig forsvarlig utvikling av moderne bioteknologi innebærer, se særlig Ot.prp. nr. 8 (1992–93) kapittel 4.12 side 45-46, kapittel 5.1 side 48-50 og kapittel 9 side 67-68. Framhevet blir blant annet naturens integritet og egenverdi som et viktig etisk prinsipp, og betydningen av offentlighet for en samfunnsmessig forsvarlig utvikling.
Begrepet bærekraftig utvikling er vanlig i norsk miljøforvaltning, men var forholdsvis nytt i lovverket. Når det gjelder framstilling og bruk av genmodifiserte organismer, gir Ot.prp. nr. 8 (1992–93) en del eksempler på hva begrepet innebærer, jamfør kapittel 5.1.3 side 50-51. Vedlegg 4 til forskrift om konsekvensutredning etter genteknologiloven gir en anvisning på hvilke elementer som kan være relevante i vurderingen av om en genmodifisert organisme bidrar til en bærekraftig utvikling. Det er basert på Bioteknologinemndas (nå Bioteknologirådet) publikasjon Bærekraft, samfunnsnytte og etikk i vurderinger av genmodifiserte organismer; Operasjonalisering av begrepene i genteknologilovens §§ 1 og 108. Det vises i forarbeidene også til Konvensjonen om biologisk mangfold9 og til den såkalte Agenda 21, handlingsplanen for det 21. århundret, vedtatt på FN-konferansen om miljø og utvikling (UNCED) i Brasil i 1992 (Ot.prp. nr. 8 (1992–93) side 9).
Hensynet til bærekraft, samfunnsnytte og etikk og EU-godkjente GMO-er er nærmere omtalt i pkt. 6.2.7.
6.2.7 Godkjenning utsetting
Paragraf 10 inneholder krav til godkjenning for utsetting. Første ledd slår fast at utsetting av genmodifiserte organismer kan bare skje etter godkjenning av Kongen. Utsetting som nevnt i § 9 bokstav a, b, c og f skal som hovedregel bare kunne skje trinnvis. Et produkt kan ikke godkjennes for omsetning før det er tilfredsstillende utprøvd i naturmiljøer som vil bli berørt av den planlagte bruk. Det kreves ikke godkjenning av annen utsetting av et produkt som er godkjent for omsetning etter denne bestemmelsen.
Genteknologiloven § 10 forutsetter en sak-til-sak-vurdering av GMO-er og det er som hovedregel ikke åpning for at forskrift kan fastsette mer generelle regler. Hovedregelen er dessuten at utsetting av GMO, som nevnt i § 9 bokstav a (feltforsøk), b (i næringsøyemed, til opprenskningsformål og lignende), c (i veksthus, akvakulturinnretning og lignende) og f (omsetning) bare skal kunne skje trinnvis. En GMO kan ikke godkjennes før den er tilfredsstillende utprøvd i naturmiljøer som vil bli berørt av den planlagte bruk. Vanligvis må ikke bare effektene i de avgrensede områder der produktet vil bli anvendt vurderes, men også mulig spredning og effektene av dette for tilgrensende områder, jamfør lovteksten og merknadene i Ot.prp. nr. 8 (1992–93) kapittel 9 side 80.
Klinisk utprøving av legemidler som består av eller inneholder GMO, anses som innesluttet bruk eller utsetting av GMO. Det vurderes fra sak til sak om reglene for innesluttet bruk eller for utsetting kommer til anvendelse, basert på legemidlet og studien. For slike legemidler er det nødvendig med tillatelse både etter legemiddelregelverket og genteknologiloven
Klinisk behandling med GMO-legemidler som er godkjent for omsetning (det vil si med markedsføringstillatelse), krever ikke tillatelse etter genteknologiloven for utsetting. Om det er krav til inneslutting ved tilvirking og administrering av det godkjente GMO-legemidlet til pasienter på sykehus, er avhengig av risikoklassen til GMO-en. Kravene til tilvirking og administrering etter genteknologiloven er beskrevet i tabell 6.1. I forbindelse med søknad om markedsføringstillatelse (omsetting) vurderer legemiddelmyndighetene kravene til tilvirking og administrering.
Det ble gjort et unntak fra godkjenningskravet etter genteknologiregelverket i både Norge og EU, for covid-vaksiner i kliniske studier og til kommersiell bruk under covid-pandemien, for å sikre rask tilgang. Dette ble hjemlet i genteknologiloven §10 femte ledd som åpner for bruk av melding.
I 2008 tok Klima- og miljødepartementet og andre berørte departementer stilling til forholdet mellom genteknologiloven og legemiddelloven når det gjaldt GMO-legemidler til mennesker og dyr. Det ble da bestemt at søknad om markedsføring av GMO-legemidler ikke lenger skulle godkjennes etter genteknologiloven, men etter legemiddelregelverket. Dette er i tråd med unntaksadgangen i utsettingsdirektivet artikkel 12. Norsk praksis for markedsføringsgodkjenning er dermed i overensstemmelse med praksis i EØS. Godkjenningen etter legemiddelregelverket, der Legemiddelverket deltar i godkjenningsprosedyren, baseres på en vurdering av om legemiddelet oppfyller kravene til kvalitet, sikkerhet og effekt og en miljørisikovurdering (sistnevnte på grunnlag av kravene i utsettingsdirektivet). Det gjøres også en miljørisikovurdering etter legemiddelregelverket. Praksis siden 2008 for behandlingen av søknader om markedsføring av GMO-legemidler har dermed vært at Legemiddelverket som myndighet etter legemiddelregelverket, har ansvar for vurdering av legemidlet gjennom EØS-prosedyren for legemiddelgodkjenning. Miljødirektoratet, etter instruks fra Klima- og miljødepartementet, følger prosedyren for høring av GMO-myndighetene angående miljørisikovurderingen av GMO-legemidlet til markedsføring. Etter denne prosedyren sender Det europeiske legemiddelbyrået (EMA) miljødelen av søknader på høring til myndighetene under utsettingsdirektivet. Miljødirektoratet gir innspill om eventuell miljørisiko og miljørisikohåndteringstiltak for GMO-legemidlet direkte til EMA, med kopi til Legemiddelverket. Innspillene fra Miljødirektoratet bygger på risikovurderinger fra Vitenskapskomiteen for mat og miljø (VKM).
EU/EØS-landene har en egen mulighet til å begrense eller forby dyrking av genmodifiserte planter godkjent etter utsettingsdirektivet eller mat- og fôrforordningen på hele eller deler av eget territorium, gjennom direktiv (EU) 2015/41210 (endringsdirektivet, «opt out»-direktivet), se også kapittel 5.8.1 for omtale av regelverket. Endringsdirektivet ble innlemmet i EØS-avtalen 13. desember 2019. Klima- og miljødepartementet har vurdert at gjennomføringen av endringsdirektivet i norsk rett ikke krever lovendring, og endringsdirektivet er dermed å anse som gjennomført i norsk rett gjennom genteknologiloven.
Endringsdirektivet endrer artikkel 26 i utsettingsdirektivet. I henhold til artikkel 26 b) kan en medlemsstat begrense eller forby dyrking av GMO enten ved å be om å bli ekskludert fra søknadens geografiske virkeområde (opt out av søknaden), eller ved å legge ned forbud mot dyrking på hele eller deler av eget territorium etter at en EU-godkjenning har funnet sted. Ved nedleggelse av forbud etter en EU-godkjenning har funnet sted, skal forbudet mot dyrking begrunnes i andre hensyn enn helse og miljø, herunder miljøpolicy, by- og distriktsplanlegging, landbruk, sosioøkonomiske virkninger med mer (listen er ikke uttømmende).
Det er ikke krav om gebyr ved søknad etter genteknologiloven.
Paragraf 10 annet ledd slår fast at utsetting av genmodifiserte organismer kan bare godkjennes når det ikke foreligger fare for miljø- og helsemessige skadevirkninger. Ved avgjørelsen skal det dessuten legges vesentlig vekt på om utsettingen har samfunnsmessig nytteverdi og er egnet til å fremme en bærekraftig utvikling.
Bestemmelsen er omtalt under punkt 6.2.5 over.
Etter § 10 tredje ledd kan Kongen ved forskrift bestemme at utsetting som nevnt i § 9 bokstav g og h kan skje uten forutgående godkjenning når nærmere fastsatte vilkår er oppfylt, for eksempel vilkår om særskilt emballasje og merking av produkter. Det kan bestemmes at slik utsetting i stedet skal være meldepliktig.
Bokstav g og h gjelder kun import og transport, ikke for eksempel omsetning, altså kommersielt salg på markedet. Import og transport er nærmere regulert i forskrift 2. september 2005 nr. 1009 om merking, transport, import og eksport av genmodifiserte organismer. Når kravene til merking og emballering etter forskriften er oppfylt, kreves som hovedregel ikke alminnelig godkjenning etter genteknologiloven § 10 første ledd for transport og import, se forskriften § 6. Alminnelig godkjenning kreves imidlertid for genmodifiserte organismer som er nevnt i forskriften § 7. Genmodifiserte organismer som er omfattet av, og oppfyller kravene i, eller er godkjent i medhold av gjeldende regler for transport av farlig gods, kan transporteres uten særskilt godkjenning etter forskriften, jamfør også § 6.
Paragraf 10 fjerde ledd slår fast at det kreves ikke godkjenning av eller melding om eksport, med mindre annet er bestemt ved forskrift gitt av Kongen.
Kapittel 3 i forskrift 2. september 2005 nr. 1009 om merking, transport, import og eksport av genmodifiserte organismer angir regler for eksport av GMO til land utenfor EØS-området, og gjennomfører Norges forpliktelser etter Cartagena-protokollen. Det kreves som hovedregel melding til, og godkjenning av importlandets myndigheter før første gangs eksport av genmodifiserte organismer for utsetting i miljøet til land utenfor EØS-området, jamfør § 15. Genmodifiserte organismer i transitt, til innesluttet bruk, til direkte bruk som mat, fôr eller videreforedling, eller til land som har angitt at import kan skje uten forhåndssamtykke, er unntatt på nærmere angitte vilkår, jamfør § 15.
Etter § 10 femte ledd kan Kongen i forskrift bestemme at bestemte typer genmodifiserte organismer kan settes ut i bestemte miljøer uten godkjenning etter første ledd første punktum. Slik utsetting skal i stedet være meldepliktig.
I Ot.prp. nr. 8 (1992–93) side 82 heter det om bestemmelsen at den tar sikte på de tilfeller der bestemte organismer er utsatt i bestemte naturmiljøer, og erfaringene viser at den aktuelle form for utsetting ikke medfører fare for helse og miljø. Bestemmelsen skal ikke brukes for å fravike prinsippet om at en GMO skal testes ut trinn for trinn.
EUs utsettingsdirektiv er som nevnt tatt inn i norsk rett. Også etter direktivet er det et begrenset handlingsrom for å gi unntak fra standardprosedyrene. Det er hensynet til helse og miljø som avgjør om en GMO skal tillates eller ikke, og som ligger under de ulike prosedyrene som er etablert. Det følger av utsettingsdirektivet artikkel 4 nr. 1 at medlemsstatene skal sikre, i overensstemmelse med føre-var-prinsippet, at det treffes alle nødvendige tiltak for å unngå uønskede virkninger på menneskers helse og miljøet som følge av utsetting eller markedsføring av GMO-er.
Med hjemmel i § 10 femte ledd er det imidlertid fastsatt forskrift av 23.oktober 2020 nr. 2124 om klinisk utprøving og utlevering av GMO-legemidler til behandling eller forebygging av covid-19, som gjennomfører likelydende EU-rettsakt om dette. Forskriften innebærer at genmodifiserte organismer under utprøving som legemiddel mot covid-19, unntas fra kravet om godkjenning etter § 10. Slike genmodifiserte organismer skal i stedet være meldepliktig. Forskriften gjelder så lenge WHO klassifiserer covid-19 som pandemi, eller til en sluttdato fastsatt av myndighetene. Sluttdatoen vil avhenge av om EU-kommisjonen anser at det foreligger en folkehelsekrise forårsaket av covid-19. Hjemmelen ble i dette tilfellet benyttet uten at det forelå konkret erfaring med utsetting av slike GMO-er i bestemte miljøer og det var ikke gjort helse- og miljørisikovurderinger i henhold til forskrift om konsekvensutredning etter genteknologiloven.
Paragraf 10 sjette ledd slår fast at det kreves ikke godkjenning for omsetning av et produkt som er godkjent for omsetning i et annet EØS-land etter reglene fastsatt i EØS-avtalen vedlegg XX punkt 25d (rådsdirektiv 2001/18/EF). Myndighetene etter loven her kan likevel forby eller begrense omsetningen dersom den etter deres syn medfører risiko for helse eller miljø, eller omsetningen for øvrig er i strid med denne lovs formål.
Gjennom EØS-avtalen er Norge som nevnt tilsluttet EUs godkjenningsprosedyrer for GMO til omsetning etter utsettingsdirektivet. En levende GMO som er godkjent for omsetning i et annet EØS-land er derfor tillatt også i Norge. Norske myndigheter kan likevel forby eller begrense omsetningen dersom den etter deres syn medfører risiko for helse eller miljø, eller omsetningen for øvrig er i strid med denne lovs formål, det vil si hensynet til etikk, samfunnsnytte eller bærekraft.
Bakgrunnen for dette er tilpasningsteksten til utsettingsdirektivet i EØS-avtalen, som lyder:
b) Artikkel 23 skal lyde:
«1. Når en avtalepart har berettiget grunn til å anta at en GMO som utgjør eller finnes i et produkt som er blitt rettmessig meldt og skriftlig godkjent i henhold til dette direktiv utgjør en fare for menneskers eller dyrs helse eller for miljøet, kan den aktuelle avtaleparten begrense eller forby bruk og/eller salg på sitt territorium av den aktuelle GMO som utgjør eller finnes i et produkt. Dersom det er tale om alvorlig risiko, skal avtaleparten påse at det blir iverksatt nødtiltak, som midlertidig oppheving eller stans av markedsføringen, herunder informasjon til offentligheten.
Avtaleparten skal gjennom EØS-komiteen umiddelbart underrette de øvrige avtaleparter om tiltak truffet i henhold til denne artikkel og angi grunnene for sin beslutning.
2. Dersom en avtalepart krever det, skal det i EØS-komiteen finne sted konsultasjoner om tiltakenes hensiktsmessighet. Avtalens del VII får anvendelse.»
c) Avtalepartene er enige om at direktivet bare omfatter aspekter i forbindelse med mulig fare for mennesker, planter, dyr og miljø. EFTA-statene forbeholder seg derfor retten til å anvende sin nasjonale lovgivning på området når helse og miljø ikke berøres, i den utstrekning lovgivningen er forenlig med denne avtale.
Samfunnsnytte er et kriterium hvor direkte og indirekte konsekvenser vurderes. Dette gjelder både økonomi og andre aspekter. I vurderingen vil man se på om det er behov i form av etterspørsel etter produktet, om produktet kan bidra til å løse et samfunnsproblem, og om det er andre alternativer som er bedre når det gjelder å løse samfunnsproblemet Videre omfattes både direkte virkninger og sekundærvirkninger.
Når det gjelder kriteriet bærekraft omfattes både naturens bæreevne, økonomisk bærekraft og sosiale forhold, herunder spørsmålet om fordeling innen og mellom land.
Av de tre tilleggskriteriene er etikk-kriteriet av særlig interesse fordi dette kriteriet også kan berettige unntak etter WTO-regelverket.
Det har ikke vært mange søknader etter utsettingsdirektivet. Til nå er seks genmodifiserte nelliklinjer til omsetning som snittede prydblomster godkjent etter fellesprosedyrene i utsettingsdirektivet, og tillatt omsatt i EØS-området, herunder Norge.
I 2014 foretok miljømyndighetene en revurdering av egen rettsforståelse når det gjelder rettslig status for GMO som var godkjent etter utsettingsdirektivet. Myndighetenes rettsforståelse hadde vært at EU-godkjente GMO ikke var tillatt å omsette i Norge uten en aktiv godkjenning fra norsk side. Av hensyn til behovet for rettsklarhet, valgte miljømyndighetene å legge seg på en ny fortolkning av genteknologiloven som innebar at GMO-ene skal anses som godkjent for omsetning i Norge, med mindre Norge har nedlagt forbud.
Norge har gjennom årene forbudt 13 GMO-produkter, jamfør forskrift 15. desember 2000 nr. 1268 om forbud mot omsetning i Norge av bestemte genmodifiserte produkter. Forskriften omfatter både tidligere forbud, og forbud etter at forskriften trådte i kraft. Det er nærmere redegjort for forbudene i kapittel 8. I tillegg ble en genmodifisert tobakk forbudt i 1997. Denne forskriften er nå opphevet.
Siste forbudsvedtak var i 2017. Regjeringen forbød da import av fire genmodifiserte planter i Norge; tre rapsplanter og én mais. Grunnen til at disse GMO-ene til mat og fôr ble behandlet etter utsettingsdirektivet og ikke etter GM mat- og fôrforordningen, var fordi søknadene ble sendt inn før GM mat- og fôrforordningen trådte i kraft. Regjeringen forbød de genmodifiserte rapsplantene til fôr og industrielle prosesser ut fra miljørisiko. Det vises til nærmere omtale i kapittel 8.
Regjeringen forbød videre den genmodifiserte maisen 1507 i industrielle prosesser og bruk som dyrefôr ut fra etiske betraktninger. De norske forbudene fra 2017 mot omsetning er så langt ikke blitt utfordret i WTO eller av EU. Se nærmere omtale av mais 1507-saken i kapittel 9.
Klima- og miljødepartementet er vedtaksmyndighet for genmodifiserte organismer omsøkt til omsetning (§ 9 f). Miljødirektoratet er delegert vedtaksmyndighet for øvrig utsetting, herunder utsetting i forskningsøyemed.
Myndigheten etter genteknologiloven § 10 første ledd til å behandle søknader om klinisk utprøving av GMO-legemidler er overført til Helse- og omsorgsdepartementet, jamfør kgl.res. 3. september 2021. Ved behandlingen av søknaden skal Legemiddelverket innhente uttalelse fra Miljødirektoratet som grunnlag for beslutningen, og i miljørisikovurderingen skal det legges avgjørende vekt på uttalelsene fra miljømyndighetene. Myndighetsoverføringen skal vurderes på ny når forslagene fra utvalget er lagt frem og nærmere vurdert.
6.2.8 Åpenhet
Paragraf 12 omhandler offentlighet og lyder som følger:
§ 12. Offentlighet
Offentleglova gjelder for saker som behandles etter denne loven. Selv om opplysninger ellers skal være offentlige etter andre og tredje ledd, gjelder unntakene i offentleglova §§ 20 og 21.
I saker om innesluttet bruk skal, uten hinder av taushetsplikt, følgende opplysninger alltid være offentlige:
a) beskrivelse av den genmodifiserte organismen, brukerens navn og adresse, formålet med bruken og bruksstedet
b) metoder og planer for overvåking og beredskap
c) vurderinger av hvilke virkninger som kan forutses.
I saker om utsetting skal opplysningene alltid være offentlige. Etter anmodning fra søker kan følgende opplysninger unntas fra innsyn hvis det dokumenteres at innsyn kan skade søkers interesser vesentlig:
a) opplysninger om fremstillings- eller produksjonsprosessen, unntatt opplysninger som er relevante for sikkerhetsvurderingen
b) opplysninger om kommersielle forbindelser mellom en produsent eller importør og søkeren eller innehaveren av godkjenningen
c) opplysninger som viser søkerens anskaffelser, markedsandeler eller forretningsstrategier
d) opplysninger om DNA-sekvenser, unntatt sekvenser som brukes til å påvise, identifisere og kvantifisere transformasjonshendelsen (genmodifiseringshendelsen)
e) opplysninger om avlsmønstre og avlsstrategier.
Andre punktum gjelder ikke risikovurderinger eller opplysninger som inngår i konklusjonene til relevante vitenskapelige utvalg eller i konklusjonene i vurderingsrapportene, og gjelder påregnelige virkninger for menneskers helse, dyrehelse eller miljøet. Departementet kan gi forskrift om at også andre opplysninger kan unntas fra innsyn i saker om utsetting.
EØS-avtalens bestemmelser om innesluttet bruk og utsetting inneholder regler om konfidensialitet og opplysninger av konkurransemessig betydning for søkeren, jamfør EØS-avtalen vedlegg XX nr. 24 og 25 d (artikkel 18 i direktiv 2009/41/EF og artikkel 25 i utsettingsdirektivet). Paragrafen ble endret ved lov 20. mai 2022 nr. 30 om endringer i genteknologiloven (offentlighet) og skiller nå mellom saker om innesluttet bruk og saker om utsetting.
Når det gjelder innesluttet bruk fastslår reglene at visse opplysninger alltid skal være offentlige. Det gjelder opplysninger om de forhold som er gjengitt i § 12 annet ledd a)–c). Adgangen til å unnta opplysninger fra offentligheten er dermed noe snevrere enn etter lov 19. mai 2006 nr. 16 om rett til innsyn i offentleg verksemd (offentleglova) § 13 sammenholdt med lov 10. februar 1967 om behandlingsmåten i forvaltningssaker (forvaltningsloven eller fvl.) § 13 første ledd nr. 2, jamfør Ot.prp. nr. 8 (1992–93) kapittel 4.8 side 40–42.
Bestemmelsene om offentlighet i saker om utsetting, bygger på Europaparlaments- og rådsforordning (EU) 2019/1381 av 20. juni 2019 (åpenhetsforordningen) som er hjemlet i General Food Law. Det fremgår av Prop. 60 LS (2021–2022) side 18–19 at bestemmelsene innebærer en innstramning i adgangen til å unnta opplysninger fra innsyn, og at myndighetene skal benytte handlingsrommet etter bestemmelsen til å gi innsyn.
Paragraf 13 gjelder offentlig høring. Den slår fast at ved saker som krever godkjenning etter denne loven, kan godkjenningsmyndigheten bestemme at det skal gjennomføres en offentlig høring. Det heter videre i bestemmelsen at det skal alltid gjennomføres offentlig høring i saker som gjelder godkjenning av søknad om utsetting av genmodifiserte organismer. Høring skal holdes i god tid før søknaden blir avgjort. Høringsprosessen må gjennomføres på en måte som sikrer at allmennheten, og i særlig grad berørte interessegrupper, får tilgang til relevant informasjon og gis en reell mulighet til å komme med synspunkter og kommentarer i saken. Beslutningen om at offentlig høring skal holdes, skal kunngjøres.
Lovbestemmelsen innebærer at det alltid skal gjennomføres offentlig høring i saker som gjelder godkjenning av søknader om utsetting (men ikke innesluttet bruk) av genmodifiserte organismer. Bestemmelsen tar sikte på å oppfylle Århuskonvensjonen artikkel 6 nr. 11, samt sikre allmennhetens deltakelse i beslutningsprosessene, jamfør Ot.prp. nr. 116 (2001–2002) side 181.
Bestemmelsen stiller nærmere krav til høringen. Plikten til høring følger også av utsettingsdirektivet artikkel 9. Tidsfristene ved utsetting av genmodifiserte organismer i genteknologiloven, jamfør innesluttet bruk-direktivet og utsettingsdirektivet, må tas hensyn til ved fastsettelsen av høringsfrister med videre, jamfør Ot.prp. nr. 116 (2001–2002) side 181.
6.2.9 Merking, sporing, deteksjon av GMO
Merking
Paragraf 14 gjelder merkeplikt. Det følger av bestemmelsen at Kongen kan gi forskrift om merking av produkter som består av eller inneholder genmodifiserte organismer eller produkter fra klonede dyr.
Merking av GMO reguleres av forskrift til genteknologiloven om merking, transport, import og eksport av GMO (forskrift 2. september 2005 nr. 1009). Paragraf 19 krever at et godkjent GMO-produkt skal merkes med at det inneholder GMO. Bestemmelsene om merking gjelder for genmodifiserte organismer som i henhold til genteknologiloven § 10 er godkjent for utsetting som definert i genteknologiloven § 9 a-f, eller som i henhold til loven § 7 er godkjent eller meldt for innesluttet bruk som definert i loven § 5, eller som kan transporteres i henhold til loven § 10 og bestemmelsene i forskriften kapittel 2.
Nærmere bestemmelser om merkingen gis i forskriften. Paragraf 19 gjelder ikke for næringsmidler, fôrvarer og såvarer, jamfør § 2, se kapittel 6.4 for omtale av disse regelverkene.
Utsettingsdirektivet regulerer merking blant annet i artikkel 19 nr. 3, artikkel 21 og vedlegg IV. Artikkel 19 nr. 3 omhandler tillatelse til markedsføring. Det fremgår her at den skriftlige tillatelsen skal i alle tilfeller uttrykkelig angi merkekrav i overensstemmelse med kravene i vedlegg IV. Merking kan altså ikke unnlates. Merkingen skal klart angi at produktet inneholder en GMO, på en etikett eller i et dokument som ledsager produktet. I vedlegg IV gis noen nærmere regler. Kravene differensieres ikke.
Artikkel 21 inneholder også noen unntak fra merkekravet, knyttet til terskelverdier av GMO.
Merking reguleres for EU-landene videre av mat- og fôrforordningen og av forordning (EF) nr. 1830/2003 om sporbarhet og merking av GMO m.m., se nærmere omtale i kapittel 6.4.2.2 og 6.4.2.3. Norge forhandler per i dag om tilpasningstekst med EFTA-landene og EU for gjennomføring av disse regelverkene, se nærmere omtale under punkt 6.4.2.2 og 6.4.2.3.
Sporing
Sporbarhet er ikke omtalt i genteknologiloven, men i forskrift om konsekvensutredning etter genteknologiloven. Vedlegg 1 Del A i forskriften beskriver opplysninger som skal gis i søknad om utsetting av andre genmodifiserte organismer enn høyerestående planter. Opplysningene skal blant annet omfatte Metoder for sporing av de genmodifiserte organismene og overvåking av virkningene, jamfør punkt V, A 1.
Den samme bestemmelsen finnes i utsettingsdirektivet Vedlegg III A om opplysninger som skal gis i søknad om utsetting av andre genmodifiserte organismer enn høyerestående planter. Opplysningene skal blant annet omfatte metoder for sporing av de genmodifiserte organismene og for overvåking av virkningene, jamfør punkt V, A 1.
Sporing er videre regulert i utsettingsdirektivet artikkel 4 nr. 6. Denne bestemmelsen forplikter medlemsstatene til å treffe tiltak for å sikre sporbarhet på alle stadier av omsetningen av GMO-er, som er tillatt for omsetning, i overensstemmelse med kravene i bilag IV.
Sporbarhet reguleres for EU-landene videre av forordning (EF) nr. 1830/2003 om sporbarhet og merking av GMO m.m., se nærmere omtale i kapittel 6.4.2.3. Forordningen innebærer blant annet at utsettingsdirektivet artikkel 4 nr. 6 erstattes av EUs regler. Forordningen er foreløpig ikke innlemmet i EØS-avtalen.
Deteksjon/påvisning
Deteksjon (påvisning) er ikke omtalt i selve genteknologiloven, men i forskrift om konsekvensutredning etter genteknologiloven. Det fremgår av forskriften vedlegg 1, som omhandler opplysninger som er nødvendige for beskrivelse av det tiltaket det søkes om godkjenning for i henhold til forskriften § 13–§ 16. Her omtales påvisning på følgende steder:
Del A Opplysninger som skal gis i søknaden om utsetting av andre genmodifiserte organismer enn høyerestående planter
II A Donor-, mottaker eller (hvis relevant) foreldreorganismens egenskaper:
…
6. Beskrivelse av identifiserings- og påvisnings
C Den modifiserte organismens egenskaper
2. Opplysninger om den endelige genmodifiserte organismen:
…
f. beskrivelse av identifiserings- og påvisningsningsteknikker for den innsatte sekvensen og vektoren,
B Vitenskapelige opplysninger
…
2. b. ii: Opplysninger om sekvensene som faktisk er satt inn eller fjernet:
-Størrelse på og antall kopier av alle påviselig innsatte sekvenser, både delvise og fullstendige, og metodene for karakterisering av dem
5. Beskrivelse av teknikker for påvisning og identifikasjon av GMHP-en
Del C: I tillegg til opplysningene i del a og b skal følgende opplysninger oppgis i konsekvensutredningen for utsetting av produkter som består av eller inneholder GMO:
7. Metoder for påvisning, identifikasjon og eventuelt mengdebestemmelse av transformasjonshendelsen, prøver av de(n) genmodifiserte organismen(e) samt kontrollprøver og opplysninger om hvor det er mulig å få tilgang til referansematerialet. Opplysninger som av fortrolighetshensyn ikke kan innføres i den offentlig tilgjengelige delen av registrene utarbeidet av Kommisjonen, skal identifiseres.
Påvisning er også omtalt i forskrift om konsekvensutredning etter genteknologiloven vedlegg 2, som inneholder prinsipper for miljørisikovurdering iht. forskriften § 13–§ 16.
Det fremgår av pkt. C.1.3.d at «Der det er mulig, skal søkeren angi omfanget av virkningen som hver utførte undersøkelse har til hensikt å påvise og begrunne dette.»
Påvisning er også omtalt i vedlegg 3, som omhandler overvåkingsplan. Det heter her at overvåkingsplanen bør blant annet
3. omfatte generell overvåking med tanke på uforutsette skadevirkninger og, om nødvendig, særskilt overvåking (i hvert enkelt tilfelle) med fokus på skadevirkninger som er påvist i miljørisikovurderingen:
a. Særskilt overvåking av et enkelttilfelle skal utføres over en tidsperiode som er tilstrekkelig lang til at såvel umiddelbare og direkte virkninger som også forsinkede og indirekte virkninger påvist i miljørisikovurderingen, kan oppdages
….
6. vurdere mekanismene for påvisning og bekreftelse av observerte skadevirkninger på menneskers helse og på miljøet, og gi innehaver av tillatelsen eller, når det er hensiktsmessig, vedkommende myndighet, mulighet til å treffe de tiltak som er nødvendige for å beskytte menneskers helse og miljøet.
Forskriften bygger på utsettingsdirektivet med vedlegg.
Sporbarhet reguleres for EU-landene videre av forordning 1830/2003, se nærmere omtale i kapittel 6.4.2.3. Forordningen er foreløpig ikke innlemmet i EØS-avtalen.
6.2.10 Overvåking
Overvåking er ikke omtalt i genteknologiloven, bortsett fra i § 12 første ledd der det fremgår at i saker om innesluttet bruk, skal opplysninger om metoder og planer for overvåking alltid være offentlige. Overvåking reguleres imidlertid av forskrift 16. desember 2005 nr. 1495 om konsekvensutredning etter genteknologiloven §§ 13, 14, 15 og 19.
Det norske regelverket bygger på utsettingsdirektivet. Selv om vedlegg 3 i forskriften omhandler overvåkingsplan, med bestemmelser om mål, generelle prinsipper, utforming og hva en overvåkingsplan bør omfatte, så er innholdet ikke nøye presisert i regelverket.
Overvåkning finner sted etter at det er gitt tillatelse til utsetting av den genmodifiserte organismen. Virksomheten som innehar tillatelsen, er ansvarlig for å gjennomføre overvåkningen og levere rapporter med resultater fra denne.
Formålet til overvåkningen er:
å bekrefte at alle antakelser i miljørisikovurderingen som gjelder forekomst og omfang av potensielle skadevirkninger av den genmodifiserte organismen eller bruken av den, er korrekte, og
å identifisere forekomsten av skadevirkninger på menneskers helse eller miljøet som skyldes den genmodifiserte organismen eller bruken av den, og som ikke ble forutsett i miljørisikovurderingen.
Det er to typer overvåkningsplaner:
Generell overvåkningsplan, utformet med tanke på å fange opp uforutsette virkninger av utsettingen
Spesifikk overvåkningplan, utformet med tanke på å overvåke spesifikke farer identifisert gjennom helse – og miljørisikovurderingen.
6.2.11 Kontroll, tilsyn, sanksjoner
Paragraf 17 første ledd gir hjemmel for Kongen til å bestemme hvem som skal føre tilsyn med gjennomføringen av genteknologiloven og vedtak i medhold av loven.
Myndigheten er lagt til Helsedirektoratet med hensyn til innesluttet bruk, jamfør forskrift 18. mars 2010 nr. 425. Miljødirektoratet har myndighet med hensyn til utsetting etter § 9, med unntak av import og omsetning av genmodifiserte organismer i næringsmidler, fiskefôr og innsatsvarer i landbruket (fôrvarer, såvarer, plantedeler med mer), som er delegert til Mattilsynet og der delegasjonen omfatter §§ 18–21 og § 24, i henhold til brev av 22. desember 2003 fra daværende Miljøverndepartementet til daværende Landbruksdepartementet (LMD) og brev 21. januar 2004 fra LMD til Mattilsynet.
Det er hittil ikke ført tilsyn etter genteknologiloven i forbindelse med gjennomføring av klinisk utprøving av GMO-legemidler.
Etter annet ledd kan Kongen gi forskrift om internkontroll og internkontrollsystemer for å sikre at krav fastsatt i eller i medhold av denne lov overholdes. Slike regler er tatt inn i den generelle internkontrollforskriften, forskrift 6. desember 1996 nr. 1127. Helsedirektoratet er delegert myndighet til å gi forskrift om internkontroll med hensyn til innesluttet bruk, jamfør forskrift 18. mars 2010 nr. 425.
Paragraf 18 slår fast at tilsynsmyndigheten har adgang til inspeksjon av ethvert sted der det foregår virksomhet som er omfattet av genteknologiloven. Tilsynsmyndigheten kan kreve å få granske dokumenter og annet materiale som kan ha betydning for dens gjøremål etter loven.
Annet materiale er ifølge Ot.prp. nr. 8 (1992–93) for eksempel prøver eller eksemplarer av genmodifiserte organismer eller cellekulturer, eller stoffer og produkter der genmodifiserte organismer inngår som bestanddel i produktet (kapittel 9 side 85). Slike prøver kan av tilsynsmyndigheten også tas med til nærmere analyse.
Paragraf 19 omhandler opplysningsplikt. Bestemmelsen slår i første ledd fast at enhver som driver virksomhet som omfattes av genteknologiloven, plikter etter pålegg fra tilsynsmyndigheten og uten hinder av taushetsplikt, å gi de opplysninger som er nødvendig for at tilsynsmyndigheten kan gjennomføre sine oppgaver etter loven. Opplysninger kan også kreves fra andre offentlige myndigheter uten hinder av den taushetsplikt som ellers gjelder.
Hva som må regnes som nødvendig, må vurderes konkret i den enkelte sak. Avhengig av omstendighetene vil opplysningsplikten imidlertid kunne omfatte både tekniske, personellmessige, økonomiske og administrative forhold, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 85. Tilsynsmyndigheten vil være bundet av de samme regler om taushetsplikt som det organet som opplysninger hentes fra, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 85–86.
Etter annet ledd skal tilsynsmyndigheten umiddelbart varsles dersom det oppstår uhell eller andre uforutsette forhold ved framstilling og bruk av genetisk modifiserte organismer.
Uforutsette forhold omfatter også kjennskap til nye opplysninger om for eksempel organismens sprednings- eller overlevelsesevne, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 86.
Varslingen fritar ikke brukeren fra å iverksette de nødvendige tiltak for å begrense skader eller ulemper etter § 21 første ledd første punktum.
Etter § 20 kan tilsynsmyndigheten gi pålegg om øyeblikkelig stans av virksomhet som utføres i strid med genteknologiloven eller vedtak i medhold av loven. Det samme gjelder dersom framstilling og bruk av genmodifiserte organismer i medhold av loven eller vedtak i medhold av loven, viser seg å medføre fare for helse- eller miljømessige skadevirkninger. Om nødvendig kan pålegg om stans gjennomføres med hjelp fra politiet.
Siden det dreier seg om levende organismer, kan stans av virksomheten innebære at organismene må destrueres.
Det er tilstrekkelig etter genteknologiloven at det kan påvises fare for skade, uten at det allerede har inntrådt skadevirkninger, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 86.
Paragraf 21 omhandler plikt til å avverge og begrense skade. Det følger av første ledd at når genmodifiserte organismer er kommet ut i miljøet i strid med genteknologiloven eller vedtak i medhold av loven, skal den ansvarlige for virksomheten treffe rimelige tiltak for å avverge eller begrense skader og ulemper. Det samme gjelder dersom genmodifiserte organismer er satt ut i miljøet i samsvar med vedtak etter loven, og det viser seg at bruken medfører større risiko for miljø eller helse enn forutsatt da bruken ble godkjent. Tilsynsmyndigheten kan pålegge den ansvarlige innen en fastsatt frist å gjennomføre oppsamling av eller treffe andre tiltak for å bekjempe organismene, herunder tiltak for å gjenopprette den tidligere tilstand i miljøet så langt det er mulig. Gjennomføring av tiltak etter denne bestemmelse kan også finne sted på annens eiendom.
Ansvarlig er ifølge Ot.prp. nr. 8 (1992–93) enhver fysisk eller juridisk person (herunder offentlig virksomhet) som driver slik virksomhet som utslippet av genmodifiserte organismer skriver seg fra. Generelt kan det sies at den som har meldeplikt eller plikt til å søke om godkjenning etter genteknologiloven, kan ilegges pålegg etter denne bestemmelse (kapittel 9 side 87).
Hva som må anses som rimelig vil bero på et skjønn i det konkrete tilfellet. Om virksomheten i utgangspunktet var lovlig vil være et viktig moment, og i tilknytning til det, kravet om forholdsmessighet.
Brukeren trenger ikke å gjennomføre et pålegg om tiltak selv, men skal sørge for at pålegget blir etterkommet innen den angitte frist, og er ansvarlig for kostnadene forbundet med det, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 88.
Annet ledd slår fast at etterkommes ikke pålegg etter første ledd innen fristen, kan tilsynsmyndigheten sørge for iverksetting av tiltakene på den ansvarliges bekostning. Det samme gjelder dersom pålegg etter første ledd kan medføre at iverksettelsen av de påkrevde tiltak forsinkes. Tilsynsmyndighetens utgifter er tvangsgrunnlag for utlegg.
Tredje ledd innebærer at krav etter § 21 foreldes 5 år etter den dag da tiltakene ble gjennomført. Dersom den ansvarlige ikke var kjent da tiltakene ble gjennomført, foreldes kravet 5 år etter den dagen da tilsynsmyndigheten fikk eller burde ha skaffet seg nødvendig kunnskap om den ansvarlige. Har tilsynsmyndigheten truffet vedtak om krav på refusjon, løper fristen fra tidspunktet da endelig vedtak ble truffet. Kravet foreldes likevel senest 30 år etter at tiltakene ble gjennomført. For øvrig gjelder reglene i foreldelsesloven så langt de passer.
Bestemmelsen fraviker foreldelsesloven for å gjennomføre Europaparlaments- og rådsdirektiv 2004/35/EF av 21. april 2004 om miljøansvar med hensyn til forebygging og utbedring av miljøskader (miljøansvarsdirektivet) artikkel 10.
Paragraf 21 a slår i første ledd fast at enhver kan anmode tilsynsmyndigheten om å gi pålegg eller treffe tiltak etter §§ 20 til 21. Tilsynsmyndigheten har plikt til å vurdere en anmodning som er fremsatt av en fysisk eller juridisk person, som berøres eller ventes å bli berørt av helse- eller miljømessige skadevirkninger, eller som har tilstrekkelig interesse i saken. Anmodningen må underbygges med opplysninger som sannsynliggjør at det foreligger helse- eller miljømessige skadevirkninger eller overhengende fare for slike.
Med tilstrekkelig interesse i saken siktes det til den samme personkretsen som etter forvaltningsloven vil ha rettslig klageinteresse, samt personkretsen med partsevne etter tvisteloven. Organisasjoner som arbeider for å fremme miljøvern er dermed omfattet.
Etter annet ledd er tilsynsmyndighetens avgjørelse på grunnlag av en anmodning som tilsynsmyndigheten etter første ledd har plikt til å vurdere, et enkeltvedtak også dersom pålegg ikke gis.
Uavhengig av utfall kan altså avgjørelsen påklages til overordnet forvaltningsorgan etter reglene i forvaltningsloven.
Paragraf 22 innebærer at Kongen kan gi forskrift om gebyrer for behandling av søknader om godkjenning etter genteknologiloven eller forskrift i medhold av loven, og for kontrolltiltak som gjennomføres for å sikre at loven eller vedtak i medhold av loven blir fulgt. Gebyrer er tvangsgrunnlag for utlegg.
Slik forskrift er ikke gitt og det er dermed ikke gebyr for søknad etter genteknologiloven.
Paragraf 23 omhandler erstatning. Det følger av første ledd at den som er ansvarlig for virksomhet etter genteknologiloven, har erstatningsansvar uten hensyn til egen skyld, når virksomheten ved utsetting eller utslipp i miljøet av genmodifiserte organismer volder skade, ulempe eller tap.
Skade omfatter både person-, tings- og formueskader. Dessuten omfattes endringer i det økologiske miljø som skjer for eksempel når en ny organisme utkonkurrerer en stedegen art (Ot.prp. nr. 8 (1992–93) kapittel 9 side 89).
Annet ledd slår fast at for øvrig gjelder reglene i forurensningsloven kapittel 8 om erstatning for forurensningsskade så langt de passer. Tilsynsmyndighetene etter genteknologiloven, trer istedenfor forurensningsmyndighetene i forurensningsloven § 58. Den som har myndighet til å gi en godkjenning etter genteknologiloven, trer istedenfor forurensningsmyndigheten i forurensningsloven § 63 annet og tredje ledd.
Erstatningsreglene tilsvarer erstatningsreglene for forurensningsskade.
Paragraf 24 omhandler tvangsmulkt. Den slår fast at ved overtredelse av vilkår, påbud eller forbud gitt med hjemmel i genteknologiloven, kan Kongen ilegge tvangsmulkt som påløper så lenge overtredelsen finner sted. Tvangsmulkten er tvangsgrunnlag for utlegg.
Myndigheten er delegert til Helsedirektoratet når det gjelder innesluttet bruk, Miljødirektoratet når det gjelder utsetting, jamfør kgl.res. 11. februar 1994 nr. 125 og forskrift 18. mars 2010 nr. 425.
Ifølge forarbeidene forutsettes det at bestemmelsen praktiseres slik at det fastsettes en frist for oppfylling av plikten, eventuelt at det når godkjenning gis, bestemmes at tvangsmulkten begynner å løpe fra en eventuell overtredelse tar til (Ot.prp. nr. 8 (1992–93) kapittel 9 side 91).
Paragraf 25 omhandler straff. Den slår fast at den som forsettlig eller uaktsomt overtrer bestemmelser gitt i eller i medhold av genteknologiloven eller vedtak truffet med hjemmel i disse bestemmelsene, straffes med bøter eller fengsel inntil 1 år.
Foreligger særlig skjerpende omstendigheter, kan fengsel inntil 4 år anvendes.
Særlig skjerpende omstendigheter er for eksempel at overtredelsen er begått grovt uaktsomt eller forsettlig, og har voldt fare for menneskers liv eller helbred eller stor miljøskade.
6.2.12 Andre bestemmelser
Paragraf 3 a gir Kongen adgang til å gi forskrift om meldeplikt ved etablering av norsk genteknologisk virksomhet i utlandet.
Bestemmelsen er blant annet en oppfølging av konvensjonen om biologisk mangfold artikkel 19.3 og 19.4, nå konkretisert gjennom Cartagena-protokollen. Det har til nå ikke vært gitt noen forskrift med hjemmel i bestemmelsen.
Paragraf 15 omhandler vilkår. I en godkjenning etter genteknologiloven §§ 6 annet ledd, 7 eller 10, eller i en dispensasjon etter § 11 b, kan det fastsettes vilkår. Slike vilkår kan gjelde blant annet valg av tekniske framgangsmåter og innsatsfaktorer som er best ut fra hensynet til helse og miljø, plikt til å tegne forsikring eller stille sikkerhet for ansvar etter §§ 21 og 23 og tiltak for å forebygge eller begrense mulige skadevirkninger. Godkjenningen kan gjøres tidsbegrenset.
Vilkår om avfallsbehandling og beredskapsplaner er andre eksempler. Tiltak for å forebygge eller begrense mulige skadevirkninger er ellers ikke begrenset til tiltak mens virksomheten foregår, men kan omfatte forebyggende tiltak i tilfelle stans eller nedleggelse av virksomheten, for eksempel forsvarlig opprydding, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 84.
Paragraf 16 omhandler endring og tilbakekall av en godkjenning. Bestemmelsen slår fast at vilkårene for en godkjenning kan endres av Kongen, og om nødvendig kan godkjenningen kalles tilbake dersom:
a. det viser seg at den aktuelle bruk medfører større risiko for helse eller miljø enn forutsatt da bruken ble godkjent, eller
b. ny teknologi gjør det mulig å minske risikoen for helse- eller miljømessige skadevirkninger, eller
c. det for øvrig følger av ellers gjeldende omgjøringsregler
Ikke enhver teknologisk utvikling kan begrunne et krav om endring eller omgjøring. Men omgjøring bør kunne finne sted dersom det innebærer en vesentlig sikkerhetsmessig gevinst. Ny teknologi omfatter også ny viten, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 85.
Gjeldende omgjøringsregler viser til forvaltningsloven § 35 og alminnelige forvaltningsrettslige prinsipper.
Paragraf 26 omhandler Bioteknologirådet. Bestemmelsen slår fast at Kongen oppnevner et råd som på begjæring eller av eget tiltak skal gi uttalelse i saker etter genteknologiloven og andre spørsmål om bioteknologi. Rådets uttalelser er offentlige, med mindre annet følger av lovbestemt taushetsplikt. Paragraf 12 gjelder tilsvarende for rådets uttalelser.
Kongen kan gi nærmere forskrifter om rådets virksomhet og sammensetning.
Bioteknologirådet ble opprettet ved kgl.res. 15. mars 1991. Bioteknologirådets mandat er ikke begrenset til gen- og celleteknologi, men omfatter bioteknologi mer generelt. Men hovedvekten av rådets virksomhet ligger i praksis på denne såkalte moderne bioteknologi. Rådet er et rådgivende organ, og har ikke myndighet til å treffe vedtak, føre kontroll eller utføre andre forvaltningsoppgaver etter genteknologiloven. Men rådet må anses som et organ for staten, jamfør forvaltningsloven § 1 annet punktum, og de øvrige reglene i forvaltningsloven vil derfor gjelde for rådets virksomhet, jamfør Ot.prp. nr. 8 (1992–93) kapittel 9 side 92.
6.3 Nærmere om GMO-legemidler
I det følgende beskrives dagens regelverk og regulering av legemidler som inneholder eller består av GMO (GMO-legemidler), først for tilstøtende legemiddelregelverk, deretter for godkjenning etter genteknologiloven. Fokus er på klinisk utprøving, men det er også inkludert noe omtale av regelverket for markedsføringstillatelse for legemidler.
6.3.1 Godkjenning etter legemiddelregelverk
6.3.1.1 Klinisk utprøving av legemidler til mennesker
Klinisk utprøving av legemidler til mennesker er regulert i forordning (EU) nr. 536/2014 om klinisk utprøving av legemidler til mennesker. Forordningen fikk anvendelse i EU 31. januar 2022, og ble vedtatt innlemmet i EØS-avtalen av EØS-komiteen 4. februar 2022. Forordningen er en stor omlegging av søknadsprosess og vurdering av kliniske utprøvinger i Europa. Regelverket med tilhørende retningslinjer er tilgjengelig på EU-kommisjonens hjemmeside i Eudralex, volum 10. Det følger av forordningen at som før, skal både legemiddelmyndigheter og etikkomiteer vurdere utprøvingen. Saksbehandlingstiden er omtrent 45 dager. Dersom myndighetene har spørsmål til søker, vil saksbehandlingstiden forlenges med 12 dager til revisjon av søknaden, pluss 19 dager til vurdering av den reviderte søknaden.
I forordning (EU) nr. 536/2014 er det en bestemmelse om en overgangsordning som ga søker mulighet til å velge om søknaden skulle sendes inn etter gammelt regelverk (direktiv 2001/20/EF), eller etter nytt regelverk (forordning (EU) nr. 536/2014 om klinisk utprøving av legemidler til mennesker). Denne bestemmelsen gjaldt til 31. januar 2023. I motsetning til tidligere regelverk, direktiv 2001/20/EF, er det nå en sentralisert innsending og vurdering av studier i EU/EØS. Søknad om gjennomføring av studier sendes til en felles EU-portal (CTIS- Clinical Trial Information System). Ved multinasjonale studier vil myndighetene i Europa samarbeide om vurderingen. Når studier skal gjennomføres i kun ett land, skal studien vurderes av det aktuelle lands legemiddelmyndighet og etikkomite. I søknaden skal søker oppgi hvilke(t) land studien ønskes gjennomført i. Det er også et samarbeid om vurderingen mellom etikkomite og legemiddelmyndighet i de enkelte landene. I Norge skal Regional Etisk Komite for Klinisk Utprøving av Legemidler og Medisinsk Utstyr, (REK KULMU) og Legemiddelverket bli enige om en felles beslutning for hver klinisk utprøving som skal gjennomføres i Norge. Det er Legemiddelverket som fatter selve vedtaket. Dersom den ene instansen er negativ til studien, skal studien ikke godkjennes i Norge. Dette må begrunnes etter gitte regler i EU-portalen (CTIS).
En klinisk utprøving gjennomføres for å finne ut hvordan et legemiddel virker, hvilke bivirkninger det har, og hvordan det omsettes i kroppen. Legemidlene som undersøkes kan både være nye legemidler og godkjente legemidler som allerede er i salg. De senere årene har Legemiddelverket årlig mottatt ca. 120 søknader om klinisk utprøving av legemidler til mennesker, mens det i 2021 var en økning til 144, og i 2022 165 søknader. De senere årene har det vært 0-5 søknader per år om kliniske utprøvinger med GMO-legemidler.
Legemidler med GMO til mennesker er gjerne legemidler til avansert terapi, og kan derfor være komplekse å vurdere. Det kan legges til 30 ekstra dager til saksbehandlingstiden for disse studiene. Normal saksbehandlingstid hos Legemiddelverket for denne typen studier varierer mellom 70–110 dager.
Sytti prosent av søknadene om kliniske studier sendes inn av et legemiddelfirma, eller en Clinical Research Organisation (CRO), som produsenten har valgt ut til å sende inn på vegne av seg. En CRO er et konsulentselskap som jobber på oppdrag fra biotech/legemiddelindustrien i forbindelse med planlegging, gjennomføring og analyse samt rapportering av kliniske utprøvninger av nye legemidler. Legemiddelfirma inngår avtaler med relevante forskningsmiljø i helsetjenesten slik at disse kan inkludere pasienter og gjennomføre studieprosedyrer i studiene.
Legemiddelmyndighetene og etikkomiteen baserer sin vurdering av om nytte/risiko-forholdet er positivt på den dokumentasjonen som søker sender inn sammen med søknaden. Søker skal blant annet sende inn dokumentasjon på erfaring fra non-kliniske studier (in vitro studier og in vivo prekliniske dyrestudier), hvordan legemidlet er produsert (det skal produseres etter såkalt god produksjonsstandard – GMP), dokumentasjon på eventuell tidligere klinisk erfaring, og en diskusjon av etiske problemstillinger. Myndighetene vurderer også om studien er planlagt gjennomført etter internasjonal god klinisk utprøvingspraksis (ICH-GCP).
Dersom legemiddelmyndighetene og/eller etikkomiteen har innsigelser til søknaden, har søker rett til å supplere søknaden én gang. Dersom søknaden ikke suppleres, eller dersom legemiddelmyndighetene, og/eller etikkomiteen til tross for endringen opprettholder innsigelsene, anses søknaden avslått og utprøvingen kan ikke iverksettes. Avslag på søknad om klinisk utprøvning skal begrunnes. Helse- og omsorgsdepartementet behandler eventuelle klager på avslag for søknader. Den nasjonale forskningsetiske komité for medisin og helsefag (NEM) involveres ved behov.
REK KULMU gjør den etiske vurderingen av studien. I tillegg til ovenfornevnte dokumentasjon, vurderer de også informasjon og samtykkeskriv, rekruttering av forsøkspersoner, utprøvers egnethet (CV), studiesentres egnethet, forsikring av forsøkspersonene, finansiering, og overholdelse av nasjonale regler vedrørende personvern og humant biologisk materiale. Dette gjelder vurdering av forskning med alle typer legemidler. REK KULMU er satt sammen av personer med ulik fagbakgrunn, lekrepresentanter og representanter for pasientforeninger. Komitéene (to stykker) oppnevnes av Kunnskapsdepartementet for fire år av gangen.
6.3.1.2 Klinisk utprøving av legemidler til dyr
Klinisk utprøving av legemidler til dyr er regulert i legemiddelloven [og i forskrift om legemidler til dyr]. Fra 28. januar 2022 kom den nye forordningen om legemidler til dyr (forordning (EU) nr. 2019/6 om legemidler til dyr) til anvendelse i EU. Endringen medførte en overgang fra direktiv til forordning. Dette betyr samme regelverkstekst i alle land, noe som vil redusere nasjonale forskjeller. Forordningen representerer hovedrettsakten på området legemidler til dyr, og er gjennomført som lov i Norge. Forpliktelsen til å etterleve den nye forordningen er derfor absolutt. Flere sekundærrettsakter får også anvendelse fra samme tidspunkt – disse gir mer detaljerte bestemmelser på enkelte punkter. Ytterligere sekundærrettsakter vil komme etter hvert. Med den nye forordningen ble det innført et overordnet krav om at klinisk utprøving skal godkjennes av relevant nasjonal myndighet før utprøvingen kan starte.
Legemiddelverket godkjenner klinisk utprøving av legemidler til dyr og har publisert en veileder for klinisk utprøving med legemidler til dyr. Retningslinjene gjelder alle typer preparater til dyr. For fiskevaksiner finnes imidlertid et eget vedlegg som bør brukes sammen med de generelle retningslinjene. Legemiddelverket mottar årlig 5-12 søknader om kliniske utprøvinger på legemidler til dyr. De fleste utprøvingene er på vaksiner og andre legemidler til fisk, men det har også vært søknader om utprøvinger på hund, geit og sau. Legemiddelverket har ikke mottatt søknader om utprøving av GMO-legemidler til dyr.
6.3.1.3 Markedsføringstillatelse
6.3.1.3.1 Markedsføringstillatelse (omsetning) for legemidler til mennesker
Legemiddelloven og legemiddelforskriften implementerer de mest relevante EU-rettsaktene, direktiv 2001/83/EF om legemidler til mennesker og forordningene (EF) nr. 726/2004 om prosedyrer for godkjenning og oppfølging av legemiddelsikkerhet til mennesker og dyr og (EF) nr. 1394/2007 om avanserte terapier for regulering av markedsføringstillatelser. Sistnevnte er et tillegg til direktiv 2001/83/EF og omhandler legemidler til avansert terapi. Et legemiddel blir bare godkjent for salg dersom legemiddelet har en nytte som overstiger risikoen ved bruk. Vurdering av nytte-/risikoforholdet til et legemiddel er basert på dokumentasjon som produsentene må sende inn når de søker om markedsføringstillatelse. I søknaden må produsenten dokumentere legemidlets farmasøytiske kvalitet, sikkerhet og medisinske effekt. Godkjenningsprosessen skjer i de fleste tilfeller i samarbeid med andre europeiske legemiddelmyndigheter, enten som en såkalt sentral prosedyre, eller som en desentralisert prosedyre.
Alle nye legemidler, inkludert GMO-legemidler, skal søkes godkjent i sentral prosedyre, slik at markedsføringstillatelsen (vilkårene for omsetting) blir lik i alle EØS-land. I forbindelse med vurdering av sikkerheten til legemidlet når det søkes om markedsføringstillatelser, vurderes også miljørisikoen for legemidlet. For legemidler som inneholder GMO skal miljørisikovurderingen utføres i henhold til kravene for miljørisikovurdering av GMO gitt i utsettingsdirektivet, og det er satt opp en GMO-prosedyre for legemidler som skal ivareta denne, beskrevet i avsnitt 6.3.1.3.3.
6.3.1.3.2 Regulering av markedsføringstillatelse (omsetning) for legemidler til dyr
Legemiddelverket har ansvar for godkjenning av legemidler til dyr. Legemiddelloven og legemiddelforskriften implementerer de mest relevante EU-rettsaktene, direktiv 2001/83/EF om legemidler til mennesker og forordning (EF) nr. 726/2004 om prosedyrer for godkjenning og oppfølging av legemiddelsikkerhet til mennesker og dyr. Godkjenningsprosessen skjer i de fleste tilfeller i samarbeid med andre europeiske legemiddelmyndigheter, enten som en såkalt sentral prosedyre, eller som en desentralisert prosedyre. En del legemidler til fisk søkes imidlertid godkjent nasjonalt i Norge. Alle nye legemidler som er fremstilt ved hjelp av bioteknologi, inkludert GMO-legemidler, skal søkes godkjent i sentral prosedyre, slik at markedsføringstillatelsen (vilkårene for omsetting) blir lik i alle EØS-land.
For andre legemidler med nytt virkestoff, eller som er innovative på annen måte, kan produsentene foreløpig velge om de vil søke i sentral prosedyre. Fra 28. januar 2022 kom et nytt EU-regelverk for legemidler til dyr til anvendelse i EU. Nå må alle legemidler med nytt virkestoff søkes i sentral prosedyre. Legemiddelmyndighetene vurderer dokumentasjon av kvalitet, sikkerhet og effekt som søker legger ved søknaden. Sikkerheten til personer som skal håndtere legemidlene, vurderes også. For legemidler til matproduserende dyr er sikkerheten til konsumenter som spiser matvarer fra behandlede dyr, et viktig aspekt.
Alle som produserer, importerer og omsetter legemidler til dyr, må også ha godkjenning fra Legemiddelverket. Legemiddelverket vurderer også søknader om forskrivning av legemidler som krever spesiell tillatelse til rekvirering.
Mattilsynet er ansvarlig myndighet for bruk av legemidlene og fører tilsyn med at dyrehelsepersonell bruker legemidler forsvarlig.
I forbindelse med vurdering av sikkerheten til legemidlet, vurderes også miljørisikoen for legemidlet. For legemidler som inneholder GMO skal miljørisikovurderingen utføres i henhold til kravene for miljørisikovurdering av GMO gitt i utsettingsdirektivet, og det er satt opp en GMO-prosedyre for legemidler som skal ivareta denne, beskrevet i punkt 6.3.1.3.3.
6.3.1.3.3 Vurdering av GMO-legemiddel ved søknader om markedsføringstillatelse (omsetning)
GMO i legemidler til både mennesker og dyr som søkes godkjent til markedsføring (omsetning) etter legemiddelregelverket i EU/EØS, behøver ikke egen godkjenning til omsetning etter utsettingsdirektivet som angitt gjennom artikkel 12 nr. 2 i utsettingsdirektivet (norsk oversettelse):
2. Når det gjelder rådsforordning (EØF) nr. 2309/93, får artikkel 13-24 ikke anvendelse på GMO-er som utgjør eller inngår i produkter dersom de er godkjent i henhold til nevnte forordning, under forutsetning av at en særskilt miljørisikovurdering utføres i samsvar med prinsippene fastsatt i vedlegg II til dette direktiv og på grunnlag av den type opplysninger som er angitt i vedlegg III, uten at det berører andre relevante krav med hensyn til risikovurdering, risikohåndtering, merking, eventuell overvåking, informasjon til allmennheten og beskyttelsesklausul som er fastsatt i Fellesskapets regelverk om legemidler for mennesker og dyr.
Forordning (EF) nr. 726/2004 om prosedyrer for godkjenning og oppfølging av legemiddelsikkerhet til mennesker og dyr speiler denne bestemmelsen i utsettingsdirektivet gjennom artikkel 6 om godkjenning av legemidler til mennesker, og artikkel 31 om godkjenning av legemidler til dyr, og angir kravene for GMO-delen av søknader om markedsføringstillatelse av GMO-legemidler. GMO-delen av søknaden skal bestå av:
Kopi av tillatelser utstedt av nasjonale kompetente myndigheter for utsetting av de genmodifiserte organismene for forsknings- og utviklingsøyemed som angitt i del B av utsettingsdirektivet
Komplett teknisk dossier som inneholder informasjonen som kreves etter vedleggene III og IV til utsettingsdirektivet
Miljørisikovurderingen i tråd med prinsippene nedfelt i vedlegg II til utsettingsdirektivet
Resultatene av eventuelle undersøkelser utført i forbindelse med forskningen og utviklingen.
Det er videre spesifisert at utsettingsdirektivet artikkel 13-24 ikke gjelder, at de vitenskapelige komiteene i sin vurdering av GMO-legemidlene skal respektere miljørisikokravene i utsettingsdirektivet, at institusjoner/organer under utsettingsdirektivet skal rådspørres ved slike søknader om markedsføringstillatelse av GMO-legemidler, og at Kommisjonen/EMA/medlemslandene skal sette opp en prosedyre som ivaretar kravene over.
Klima- og miljødepartementet og andre berørte departementer besluttet i 2008 at det i tråd med EU-regelverket ikke behøves egen godkjenning etter genteknologiloven for søknader om markedsføringstillatelse (omsetning) av GMO-legemidler. Det er tilstrekkelig at disse godkjennes av legemiddelmyndighetene etter legemiddelloven. Ved søknad om markedsføringstillatelse gjør legemiddelmyndighetene en risiko/nytte-vurdering og en miljørisikovurdering av legemidlet, etter legemiddelregelregelverket, og ikke etter genteknologiloven. Norske GMO-myndigheter i likhet med GMO-myndighetene etter utsettingsdirektivet i EU, spiller imidlertid inn i den felles prosedyren i EU, som omtales i neste avsnitt. Klima- og miljødepartementet har foreslått en endring av genteknologiloven som skal ta inn denne praksisen.
Praksis siden 2008 for behandlingen av søknader om markedsføring av GMO-legemidler har dermed vært at Miljødirektoratet, etter instruks fra Klima- og miljødepartementet, følger EØS-prosedyren for høring av GMO-myndighetene om miljørisiko og miljørisikohåndtering ved søknad om markedsføringstillatelse av GMO-legemidler. GMO-prosedyren hvor GMO-myndighetene blir rådspurt i forbindelse med søknad om markedsføringstillatelse av GMO-legemidler koordineres av det europeiske legemiddelbyrået, EMA. Søker legger ved en GMO-del i henhold til kravene i utsettingsdirektivet for vurdering av de(n) genmodifiserte organismene, og det oppnevnes/meldes i forbindelse med behandlingen av GMO-delen av søknaden en rapportør (og co-rapportør) som utarbeider en risikovurderingsrapport for GMO-en i legemidlet. GMO-myndighetene får først tilsendt den tekniske informasjonen om GMO-en i henhold til kravene i vedlegg III og IV i utsettingsdirektivet (såkalt modul 1.6.2), en miljørisikovurdering i tråd med vedlegg II i utsettingsdirektivet, og dokumentasjon og resultater av tidligere forsøk i forbindelse med utviklingen av GMO-en. Deretter får myndighetene tilsendt rapportørrapportene utarbeidet for GMO-delen. GMO-myndighetene spiller inn i en felles innspillsrunde til en gitt frist satt av EMA. Innspill vurderes av EMA/legemiddelkomiteene, som fremmer eventuelle spørsmål til søker samlet for legemidlet. Innspillene fra GMO-myndighetene er konsentrert omkring identifikasjon av spesifikke risikoer for spredning av GMO-en og håndtering av disse gjennom kontrolltiltak. Kontrolltiltakene skal angis som håndteringsinstrukser til det godkjente GMO-legemidlet.
6.3.2 Godkjenning av GMO-legemiddel i klinisk utprøving etter genteknologiloven
Når legemidlene inneholder levende genmodifiserte organismer, vurderes legemiddelutprøvingen også etter genteknologiloven. Framstilling og bruk av GMO i klinisk utprøving kan godkjennes som innesluttet bruk og/eller utsetting, som oftest basert på oppsettet til studien, og/eller GMO-ens egenskaper.
Godkjenningsmyndigheter etter genteknologiloven for innesluttet bruk er Helsedirektoratet, og Legemiddelverket for utsetting av GMO i klinisk utprøving av legemidler (se nærmere omtale under i 6.3.2.2). Det fremgår av kgl.res. 3. september 2021 side 2 tredje avsnitt at ordningen skal gjelde «frem til forslagene fra GMO-utvalget er lagt frem og nærmere vurdert». Vedtaksmyndighetene er de samme, uavhengig av om GMO-legemidlene er til mennesker eller dyr.
6.3.2.1 Innesluttet bruk
Genteknologiloven § 5 definerer innesluttet bruk som (…) enhver arbeidsoperasjon hvor genmodifiserte organismer blir framstilt, dyrket, lagret, destruert eller brukt på annen måte, i et lukket system hvor det anvendes fysiske inneslutningstiltak, eventuelt i kombinasjon med andre særskilte inneslutningstiltak, for å begrense organismenes kontakt med mennesker og miljø slik at disse sikres et høyt nivå av sikkerhet. Det fremgår av § 6 første ledd at innesluttet bruk skal skje i laboratorier og anlegg som er godkjent etter annet ledd, og i samsvar med god mikrobiologisk praksis.
Etter loven er all bruk av en genmodifisert organisme som ikke skjer i et lukket system som nevnt i § 5, å anse som utsetting. Praksis de senere år har derfor vært å anse klinisk utprøving av GMO-legemidler til mennesker som utsetting, men at håndtering av GMO-legemiddelet skjer under innesluttet bruk-reglene for å sikre beskyttelse mot eventuell eksponering av andre pasienter i helseinstitusjon hvor medikamentet gis. Regelverk og prosedyre for utsetting omtales i 6.3.2.2.
Fremstilling og bruk av genmodifiserte mikroorganismer er regulert gjennom egen forskrift til genteknologiloven; forskrift om innesluttet bruk av genmodifiserte mikroorganismer. Det er fire inneslutningsnivåer for genmodifiserte mikroorganismer, 1-4, hvor inneslutningsnivå 1 er det minst strenge. Inneslutningsnivåene er basert på risikokategorisering av mikroorganismene, nærmere bestemt sykdomsfremkallende evner og evne til å overleve og spre seg utenfor de innesluttede fasilitetene.
Inneslutningsnivå til testfasiliteter for utprøving av GMO-legemidler til mennesker vurderes fra sak til sak av Helsedirektoratet, men Helsedirektoratet har som generelt utgangspunkt for mottak, tilvirking og avhending av GMO-legemiddel vurdert at for genmodifiserte virus som ikke er replikasjonskompetente, vil det være tilstrekkelig med inneslutningsnivå 1. For noen GMO-legemidler med virusvektorer som replikerer i verten, for eksempel for onkolytisk virus eller andre replikasjonskompetente virus, brukes inneslutningsnivå 2 ved tilvirkning av GMO-legemiddelet. Minimumskravene til inneslutningsnivåene som angitt i forskriften er gjengitt i tabell 1 under. Det kan være andre krav til inneslutningstiltak på hvert nivå for bruk av genmodifiserte mikroorganismer i kombinasjon med dyr, og for storskala produksjonsanlegg for genmodifiserte mikroorganismer.
Tabell 6.1 Oversikt over minimumskrav til laboratorier/anlegg for innesluttet bruk av genmodifiserte mikroorganismer (GMM)
Minimumskrav for inneslutningsnivå 1 | Minimumskrav for inneslutningsnivå 2 | Minimumskrav for inneslutningsnivå 3 | Minimumskrav for inneslutningsnivå 4 |
|---|---|---|---|
Overflater (arbeidsunderlag) skal tåle vann, syrer, alkaliske stoffer, løsemidler, desinfeksjonsmidler, dekontamineringsagenser, og være lette å rengjøre | Overflater (arbeidsunderlag) skal tåle vann, syrer, alkaliske stoffer, løsemidler, desinfeksjonsmidler, dekontamineringsagenser, og være lette å rengjøre | Overflater (arbeidsunderlag, gulv) skal tåle vann, syrer, alkaliske stoffer, løsemidler, desinfeksjonsmidler, dekontamineringsagenser, og være lette å rengjøre | Overflater (arbeidsunderlag, gulv, tak, vegger) skal tåle vann, syrer, alkaliske stoffer, løsemidler, desinfeksjonsmidler, dekontamineringsagenser, og være lette å rengjøre |
Autoklav (for sterilisering) på stedet | Autoklav (for sterilisering) i bygningen | I hele laboratorieområdet1 | Laboratoriet = med gjennomgang |
Begrenset adgang kreves ikke | Adgangskontroll | Adgangskontroll | Adgangskontroll |
Brukerne skal benytte egnet vernetøy (arbeidstøy) | Egnet vernetøy (valgfritt fottøy) | Egnet vernetøy | Fullstendig kles- og fottøyskift før en går inn og ut av området |
Inaktivering av GMM i kontaminert materiale og avfall kreves | Inaktivering av GMM i kontaminert materiale og avfall kreves | Inaktivering av GMM i kontaminert materiale og avfall kreves | Inaktivering av GMM i kontaminert materiale og avfall kreves |
Inaktivering av GMM i avløpsvann fra håndvask eller avløp og dusjer og lignende spillvann: Kreves ikke | Inaktivering av GMM i avløpsvann fra håndvask eller avløp og dusjer og lignende spillvann: Kreves ikke | Inaktivering av GMM i avløpsvann fra håndvask eller avløp og dusjer og lignende spillvann: Valgfritt | Inaktivering av GMM i avløpsvann fra håndvask eller avløp og dusjer og lignende spillvann: Kreves |
Kontroll med aerosol under prøvetaking, tilførsel av materiale til et lukket system eller overføring av materiale til et annet lukket system: Valgfritt | Kontroll med aerosol under prøvetaking, tilførsel av materiale til et lukket system eller overføring av materiale til et annet lukket system: Kreves, redusere spredning mest mulig | Kontroll med aerosol under prøvetaking, tilførsel av materiale til et lukket system eller overføring av materiale til et annet lukket system: Kreves, hindre spredning | Kontroll med aerosol under prøvetaking, tilførsel av materiale til et lukket system eller overføring av materiale til et annet lukket system: Kreves, hindre spredning |
Effektiv vektorkontroll (f.eks. for å oppdage gnagere og insekter): Valgfritt | Effektiv vektorkontroll (f.eks. for å oppdage gnagere og insekter): Kreves | Effektiv vektorkontroll (f.eks. for å oppdage gnagere og insekter): Kreves | Effektiv vektorkontroll (f.eks. for å oppdage gnagere og insekter): Kreves |
1 Med validerte fremgangsmåter som muliggjør sikker overføring av materiale til en autoklav utenfor laboratoriet, og som gir et tilsvarende vernenivå.
Fremstilling og bruk av genmodifiserte mikroorganismer i laboratorier/fasiliteter som allerede er godkjent for innesluttet bruk, krever kun melding (ikke full søknad) til Helsedirektoratet. En slik melding skal sikre at virksomhetene og myndighetene er samstemte om at den aktuelle genmodifiserte mikroorganismen arbeides med på riktig inneslutningsnivå. Saksbehandlingstiden for godkjenning av fasiliteter og melding er fastsatt i regelverket og er 45 dager. Helsedirektoratet har for de fleste meldinger besvart disse raskt og aldri utover fristen på 45 dager.
Inneslutningstiltakene skal sikre at genmodifiserte mikroorganismer ikke slippes ut i miljøet og potensielt skader mennesker og miljø, og at virksomhetene til enhver tid har kontroll med hvilke genmodifiserte mikroorganismer det arbeides med i anleggene. Myndighetene skal også utarbeide beredskapsplaner for innesluttet bruk, for håndtering av uhell som medfører utslipp av genmodifiserte mikroorganismer som kan føre til alvorlig fare for mennesker og dyr og miljøet utenfor anlegget.
6.3.2.2 Utsetting av GMO gjennom klinisk utprøving av GMO-legemidler fra november 2021
Som omtalt over i 6.3.2 ble vedtaksmyndighet etter genteknologiloven for godkjenning av søknader om klinisk utprøving av GMO-legemidler til utsetting, overført fra Miljødirektoratet til Legemiddelverket 15. november 2021. Overføringen av vedtaksmyndighet gjelder både klinisk utprøving av GMO-legemidler til mennesker og til dyr.
Formålet med overføringen, samt Miljødirektoratets rolle vedrørende miljørisikovurdering, ble i kongelig resolusjon av 3. september 2021 beskrevet på følgende vis; (v)ed siden av å gjøre søknadsprosedyrene klarere og enklere for søkerne, kan dette være et grep for å få ned saksbehandlingstiden i disse sakene. Ved godkjenning etter genteknologiloven skal det innhentes uttalelse fra Miljødirektoratet som grunnlag for beslutningen, og i miljørisikovurderingen skal det legges avgjørende vekt på uttalelsene fra miljømyndighetene.
Miljørisikovurderingen innebærer å vurdere risiko for negative virkninger på mikroorganismer, dyr, planter, eller mennesker utover pasienten(e) som får legemidlet, og eventuelle kontrolltiltak for å hindre unødvendig spredning av den genmodifiserte organismen i miljøet.
Etter forskrift om konsekvensutredning etter genteknologiloven er det anledning til å ha en saksbehandlingstid på inntil 90 dager, og med mulighet for 30 dagers utvidelse ved høring (totalt 120 dager). Som nevnt over, er saksbehandlingstiden etter legemiddelregelverk om lag 45 dager, med noe tillegg. Samtidig kan søknader med GMO-legemidler ha lengre frister hjemlet i legemiddelregelverket sammenlignet med legemidler uten GMO, da vurdering av GMO-aspektene kan være komplekse og tidkrevende. Det etterstrebes å ha lik behandlingstid for vurdering etter genteknologiloven og vurderingen etter legemiddelregelverket. Legemiddelverket har derfor lagt seg på et nivå der vedtak etter genteknologiloven i utgangspunktet skal fattes innen dag 55.
Legemiddelverket og Miljødirektoratet har utarbeidet saksbehandlingsrutiner for etatene. Det er utarbeidet én rutine for utprøving av GMO-legemidler til mennesker, og en annen for utprøving av veterinære GMO-legemidler. Begge rutinene legger opp til at vedtak etter genteknologiloven ikke skal komme senere enn de fastsatte fristene i legemiddelregelverket, og dermed ikke forsinke igangsetting av studien. For GMO-legemidler til dyr har Miljødirektoratet 45 kalenderdager til å uttale seg, mens for GMO-legemidler til mennesker er Miljødirektoratet gitt en frist på 10 kalenderdager. Vitenskapskomiteen for mat og miljø (heretter kalt VKM) gis i oppdrag å vurdere miljørisiko for klinisk utprøving av GMO-legemidler, både til mennesker og dyr. Miljødirektoratet er fortsatt vedtaksmyndighet for utsetting av genmodifiserte organismer i forskningsøyemed til andre formål enn klinisk utprøving av GMO-legemidler.
Søknader om vurdering av GMO-legemiddel i klinisk utprøving av legemidler til mennesker og dyr sendes til Legemiddelverket via den felles nasjonale postkassen for kliniske studier med GMO-legemidler. Det anbefales at søker benytter Eudralink for trygg innsendelse av søknaden. Legemiddelverket har publisert en veiledning til søkere som ligger på hjemmesiden til Legemiddelverket.
Søknaden skal inneholde informasjon som gitt i konsekvensutredningsforskriften (etter genteknologiloven) vedlegg 1 del A, en helse- og miljørisikovurdering, dokumentasjon som underbygger søkers konsekvensutredning, og en SNIF (summary notification format) som er et EU-standard format for en kortfattet oppsummering av søknaden uten fortrolig informasjon. For en del produkttyper av GMO-legemidler kan det benyttes søknadsskjema utviklet gjennom GMO-interplay samarbeidet i EU. Disse søknadsskjemaene omtales nærmere i kapittel 12, og kan benyttes i de aller fleste land i Europa.
Søkere som allerede har søkt/er i ferd med å søke godkjenning i andre land etter utsettingsdirektivets del B (utsetting for andre formål enn omsetning), kan benytte denne til å søke godkjenning av utsetting av GMO i klinisk utprøving etter genteknologiloven.
Søknad om utsetting kunngjøres, jamfør genteknologiloven § 13 om offentlig høring, på Legemiddelverkets nettsider. SNIF vedlegges da høringen. Legemiddelverket tar sikte på en generell høringstid på 15 dager, både for GMO-legemidler til mennesker og dyr.
Helse- og miljørisikovurderingen av genmodifiserte organismer i legemidler følger samme metodologi for vurdering av risiko som for GMO til andre bruksområder, og er beskrevet i forskrift om konsekvensutredning etter genteknologiloven.
Konsekvensutredningen fra søker kan forelegges Bioteknologirådet for uttalelse før vedtak treffes, jamfør forskriften § 11. I motsetning til søknader om godkjenning av omsetning av GMO-legemidler etter genteknologiloven, er praksis for søknader om kliniske studier med GMO-legemidler til mennesker og dyr at de ikke forelegges Bioteknologirådet til særskilt uttalelse, men at rådet har anledning til å gi innspill om vurdering av bærekraft, samfunnsnytte og etikk i høringene av søknadene.
For GMO-legemidler som allerede innehar markedsføringstillatelse i EU/EØS, men hvor de skal prøves ut i nye kliniske studier, følger Legemiddelverket prosedyren11 utarbeidet gjennom interplay-samarbeidet i EU, hvor sponsor (søker) sender inn et skjema med informasjon om planlagte studier, og om miljørisikoen er vesentlig endret eller ikke for de nye planlagte studiene sammenlignet med den opprinnelige markedsføringstillatelsen til GMO-legemidlet.
6.4 Matlovsområdet – regelverk for genmodifisert mat og fôr mv.
Någjeldende regelverk på matområdet er i stor grad gjennomført EØS-regelverk. EUs regelverk om genmodifisert mat og fôr er foreløpig ikke tatt inn i EØS-avtalen og norsk rett, men nasjonalt regelverk for GM mat og fôr er utarbeidet på basis av EUs reguleringer på området.
6.4.1 Regelverksutvikling i EU – matområdet
EU reformerte sin matlovgivning på begynnelsen av 2000-tallet. Forordningen med kortnavn General Food Law12 (matlovsforordningen), er siden 2002 det overordnede rammeverket for alt regelverk om mat og fôr både på unionsnivå og i de enkelte medlemsstatene. Matlovsforordningen er tatt inn i norsk rett gjennom matlovsforskriften13. Food Law fastsetter allmenne prinsipper og krav til mat- og fôrregelverk i et helkjedeperspektiv og sikrer derved et høyt beskyttelsesnivå med hensyn til helse, miljø og forbrukerinteresser. Forordningen har egne bestemmelser om risikoanalyse og føre-var-prinsippet, og det er opprettet et eget informasjonsutvekslings- og varslingssystem for helsefarlig mat og fôr (Rapid Alert System for Food and Feed – RASFF14) samt risikovurderingsorganet European Food Safety Authority (heretter EFSA). Med disse harmoniserte kravene, legges det også til rette for et effektivt indre marked.
En rekke ulike faktorer kan ha en direkte eller indirekte innvirkning på mattryggheten. Food Law’s virkeområde går derfor helt fra primærproduksjonen av mat og fôr og frem til overlevering av maten til forbrukerne – «from farm to fork». Innenfor primærproduksjon vil blant annet dyrehelse, såvarer, gjødsel og plantevernmidler være omfattet, og forordningen berører også spørsmål knyttet til dyrevelferd, miljø og plantehelse.
Før EU fikk et samlet regelverk for genmodifisert mat og fôr i 2003, var det ulike regelverk som regulerte ulike bruksområder for den samme GMO. Det var ingen felles regler verken for godkjenning, merking eller sporbarhet. Både levende GMO og prosessert (synonym: ‘bearbeidet’, ‘avledet’) GMO til bruk som mat, ble godkjent i henhold til forordning (EF) nr. 258/97 om Ny mat, og levende GMO til bruk som fôr ble godkjent etter direktiv 2001/18/EF om utsetting av genmodifiserte organismer i miljøet (heretter utsettingsdirektivet). Det var ingen godkjenningskrav for prosessert genmodifisert fôr. Figur 6.1 viser skjematisk og overordnet den historiske utviklingen av krav som ble stilt i EUs regelverk for genmodifiserte produkter (unntatt legemidler). Utviklingen viser at det har blitt godkjennings- og merkekrav for stadig flere produktgrupper og at regelverkene er samlet etter sektorprinsippet.
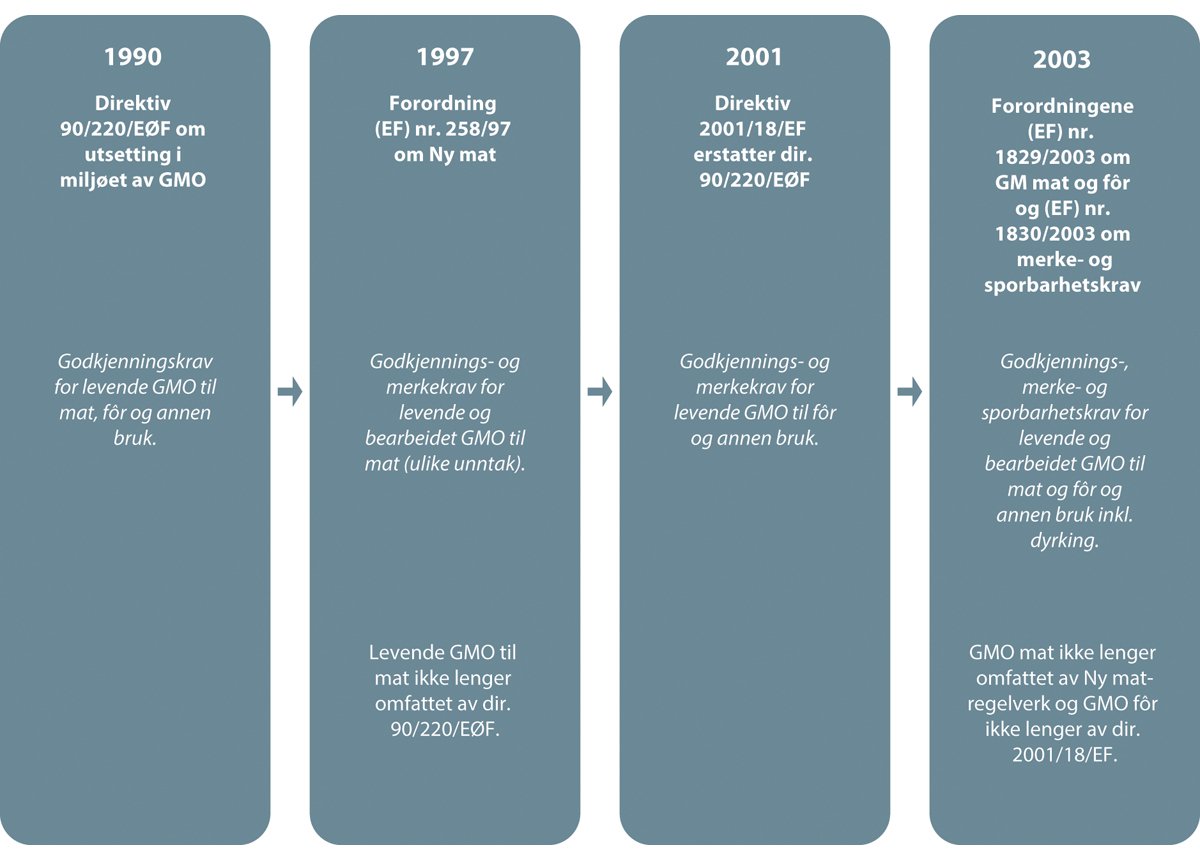
Figur 6.1 Utviklingen av EUs regelverk for genmodifiserte produkter (unntatt legemidler) med hensyn til krav om godkjenning og merking mv.
Fra 1999 nektet flere medlemsstater generelt å godkjenne GMO til bruk som mat og fôr før man fikk på plass nye fellesskapsregler for helserisikovurdering, merking og sporbarhet. Perioden frem til nytt regelverk ble vedtatt høsten 2003, kalles moratorium-perioden fordi ingen GMO ble godkjent i denne perioden. Det førte til at USA, Canada og Argentina gikk til WTO-tvistesak, fordi de mente EU hindret handel i strid med WTO-regelverket. EU tapte tvistesaken15, men laget siden et stort og omfattende regelverk for genmodifisert mat og fôr. Mer informasjon om tvistesaken finnes i kapittel 5.4.4.
6.4.2 EUs regelverk om genmodifisert mat og fôr
EUs regelverk om genmodifisert mat og fôr skal sikre trygge produkter og en enhetlig og helhetlig vurdering av GMO til bruk som mat og fôr etter ‘one door – one key’ prinsippet.
6.4.2.1 Innledning
Helhetstankegangen i General Food Law ble også brukt i utformingen av EUs nye regelverk om genmodifisert mat og fôr som ble vedtatt i 2003 og trådte i kraft våren 2004. Regelverket skal beskytte human- og dyrehelse, miljøet og forbrukerinteresser, det skal sikre et effektivt indre marked og ikke være handelshindrende. Regelverket er hjemlet i EUs Food Law, og består av 12 rettsakter – den såkalte GM-pakken. Regelverkspakken er ennå ikke innlemmet i EØS-avtalen.
Forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr16 og forordning (EF) nr. 1830/2003 om sporbarhet og merking av genmodifiserte organismer og om sporbarhet av mat og fôr fremstilt på grunnlag av genmodifiserte organismer, og endring av direktiv 2001/18/EF (sporbarhets- og merkeforordningen)17, er basisrettsaktene. De andre rettsaktene18 er hjemlet i en av de to basisrettsaktene og omhandler blant annet særskilte implementeringsregler, krav til søkers dokumentasjon, opprettelsen av et system for unike koder, krav til EUs referanselaboratorier og aksept av sporforurensninger av GMO i fôr for GMO som er omsøkt, men ennå ikke godkjent. Veilederen omhandler prøvetakings- og analysemetoder.
Rettsaktene i GM-pakken legger til rette for en enhetlig og helhetlig saksbehandling av søknader om godkjenning av GMO til bruk som mat og fôr, med basis i vitenskapelige helse- og miljørisikovurderinger. Det skilles ikke mellom levende, formeringsdyktige GMO og prosesserte eller bearbeidede produkter fra GMO. Virksomheter sender inn én søknad for alle bruksområder etter prinsippet ‘one door – one key’.
Regelverket omfatter alle GMO som forventes å kunne inngå i mat- og fôrkjeden, mens utsettingsdirektivet19 omfatter dyrking, import og omsetning av GMO som ikke forventes å inngå i mat og fôrkjeden. De senere år er det kun søknader om import og omsetning av ulike GMO-nelliker som er godkjent under direktivet. Utsettingsdirektivet utgjør likevel en av basisrettaktene på området, fordi det definerer ‘genmodifisert organisme’ og fordi kravene til miljørisikovurdering av GMO som søkes godkjent etter GM mat- og fôrforordningen er regulert der.
6.4.2.2 Forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr
Denne forordningen (GM mat- og fôrforordningen) regulerer kravene til godkjenning og merking av GMO til import, dyrking og prosessering til bruk som mat og fôr. Forordningen henviser til utsettingsdirektivet for definisjonen av GMO. Hovedelementene i forordningen fremgår nedenfor.
Godkjenningskrav. Forordningen legger opp til en sentralisert godkjenningsprosedyre som innebærer godkjenning basert på en uavhengig, vitenskapelig helse- og miljørisikovurdering foretatt av EFSA med påfølgende votering i den faste komitéen for genmodifisert mat og fôr (Standing Committee on Plants, Animals, Food and Feed, Section GM Food and Feed: SCPAFF GMFF).
Det følger av forordningen at genmodifiserte produkter ikke kan markedsføres før de er risikovurdert og godkjent, og de kan ikke omsettes uten at de er merket som genmodifisert. En søknad om godkjenning kan omfatte all bruk, dvs. dyrking, import, bruk som mat og fôr og annen bruk enn mat og fôr (for eksempel import av råvare for prosessering til emballasje, papir, råmateriale til tekstiler mv.). En søknad kan også omfatte all bruk unntatt dyrking, og det er dette som er praksis i EU i dag.
Det er ikke mulig å søke om godkjenning for bruksområdet kun dyrking av GMO til mat og fôr, eller kun oppdrett av GMO produksjonsdyr. En slik søknad kan imidlertid fremmes etter utsettingsdirektivet, men antas å ha begrenset verdi da GMO-avlingen eller GMO-dyret ikke kan brukes direkte som mat og fôr og heller ikke kan prosesseres til andre produkter uten etter godkjenning under GM mat- og fôrforordningen. Det er likevel mulig å eksportere avlingen eller GMO-dyret til land utenfor EØS. I praksis sendes alle søknader om godkjenning av GMO til bruk som mat og fôr, under GM mat- og fôrforordningen, som er hjemlet i Food Law.
Unntak. Det følger av GM mat og fôrforordningen at den ikke omfatter produkter som er fremstilt ved hjelp av GMO. Denne kategorien av produkter defineres ikke som GMO, og det er derfor verken godkjennings- eller merkekrav til produktene. Et typisk eksempel er et enzym produsert ved at det settes inn et gen som koder for proteinet i en mikroorganisme som deretter oppformeres i et innesluttet system. Enzymet renses deretter ut slik at det ikke er noe materiale fra den genmodifiserte mikroorganismen til stede i sluttproduktet. Det er krav i regelverket om at det ikke skal være noen rester fra den genmodifiserte mikroorganismen igjen i sluttproduktet for at det skal unntas godkjennings- og merkekrav. Produksjon av ulike typer enzymer og vitaminer ved hjelp av genmodifiserte mikroorganismer er en viktig industri, og er mye brukt i mat- og fôrproduksjon. Produkter fra dyr som er fôret med genmodifiserte fôrvarer eller behandlet med genmodifiserte medisiner, er andre produktgrupper som er produsert ved hjelp av GMO.
Merkekrav. Forordningen regulerer merkekrav, som er viktig for å gi forbrukerne bedre muligheter til å ta informerte valg. Merkekravene gjelder den aktuelle GMO, produkter som inneholder GMO-en og alle prosesserte produkter fra GMO-en inkludert produkter som er så prosessert at de ikke inneholder noen rester av DNA eller proteiner (prosessmerking).
Det skal klart fremgå av merkingen at produktet eller ingrediensen er genmodifisert eller fremstilt fra en genmodifisert organisme. Dersom produktet ikke er emballert, pakningen er veldig liten, eller fôrmiddelet (fôrråvaren) ikke kan merkes direkte, skal merkingen fremgå tydelig i umiddelbar nærhet til der produktet frembys (mat) eller i et ledsagende dokument (fôr). Det er videre bestemmelser om å merke særskilt hvis produktet er annerledes enn et tilsvarende ikke-genmodifisert produkt med hensyn til sammensetning og næringsverdi, eller om produktet er påtenkt en spesiell anvendelse eller visse grupper i befolkningen eller visse dyrearter.
Unntak. Mat og fôr med innhold av sporforurensninger av godkjent GMO eller prosessert GMO på maksimalt 0,9 % på ingrediensnivå, er unntatt fra merkekravene forutsatt at tilstedeværelsen er utilsiktet eller teknisk uunngåelig og at frambyder kan dokumentere dette.
Beslutningsprosessen. For å få godkjent en søknad om GMO og produkter fra GMO, må søker sende inn opplysninger om de endringer som er gjort i organismen og hvordan dette har endret dens egenskaper. Det må også sendes inn dokumentasjon som viser at den er like trygg for menneskers, dyrs og planters helse og for miljøet, som den konvensjonelle varianten den er sammenlignet med.
GM mat- og fôrforordningen er rettslig grunnlag for forordning (EU) nr. 503/2013 om søknader om godkjenning av GMO mat og fôr. Denne rettsakten stiller formelle krav til søker og hva slags dokumentasjon som kreves for at EFSA skal kunne gjennomføre en helse- og miljørisikovurdering. Søker må blant annet også levere opplysninger om analyseverktøy og prøvemateriale slik at myndighetene senere kan utøve kontroll. EFSA oversender opplysningene og prøvemateriale til EUs referanselaboratorium for validering av analysemetoden.
EFSA sender alle søknader under GM mat- og fôrforordningen på en tre måneders vitenskapelig høring til medlemsstatene og EFTA-landene. Når EFSA har ferdigstilt og offentliggjort en risikovurdering, sendes den på en 30 dagers offentlig høring, før risikovurderingen presenteres for medlemsstatene i SCPAFF GMFF. Deretter voterer medlemsstatene over EU-kommisjonens forslag. Norge har uttalerett og kan stille spørsmål til EFSA i SCPAFF GMFF, men deltar ikke under selve voteringen. Blir det ikke en flertallsbeslutning for eller imot kommisjonens forslag om godkjenning, blir det en ny drøftings- og voteringsrunde i «Appeal Committee» (ankekomitéen). Blir det ingen flertallsbeslutning i denne komitéen heller, sendes saken tilbake til EU-kommisjonen, som da er forpliktet til å godkjenne sitt eget forslag om godkjenning. Til nå er ingen søknader om godkjenning av GMO til mat og fôr, godkjent ved en flertallsbeslutning i EU.
6.4.2.3 Forordning (EF) nr. 1830/2003 om sporbarhets- og merkekrav
Sporbarhet- og merkeforordningen gir bestemmelser om sporbarhet og merking av GMO godkjent under GM mat- og fôrforordningen eller under utsettingsdirektivet (uavhengig av bruksområde). Den gir også bestemmelser om sporbarhet av produkter produsert fra GMO som skal brukes til mat og fôr.
Sporbarhet defineres i forordningen som muligheten for å spore GMO og produkter fremstilt fra GMO på alle stadier i markedsføringen og gjennom hele produksjons- og distribusjonskjeden. Hensikten er blant annet å kunne trekke tilbake fra markedet produkter som er helse- eller miljøskadelige. Sporbarhets- og merkeforordningen hjemler en rettsakt som fastsetter systemet for tildeling av unike koder20 for den enkelte GMO som godkjennes under GM mat- og fôrforordningen eller utsettingsdirektivet.
I det første leddet i markedsføringen skal virksomhetene sikre at det skriftlig gis opplysninger om at produktet består av eller inneholder GMO. Det skal videre gis skriftlig informasjon til mottakende virksomhet om at produktet inneholder eller består av GMO med den/de unike koder som ble tildelt i forbindelse med godkjenningen etter GM mat- og fôrforordningen eller utsettingsdirektivet. Virksomhetene skal også sikre at produktene er riktig merket med at produktet inneholder eller består av GMO. Opplysningene skal videre gis skriftlig til alle etterfølgende virksomheter i omsetningskjeden, og virksomhetene skal ha systemer for å oppbevare informasjon om GMO-produkter ett ledd tilbake i kjeden og ett ledd frem, i en periode på fem år.
Tilsvarende krav til sporbarhet som for levende GMO, gjelder for produkter til mat og fôr fremstilt fra levende GMO. Opplysningene skal angi hver av matvareingrediensene som er fremstilt fra GMO og hver av fôrvarene eller tilsetningsstoffene som er fremstilt fra GMO. For produkter der det ikke er noen ingrediensliste, skal det opplyses om at produktet er fremstilt fra GMO. Virksomhetene skal ha systemer som sikrer sporbarheten av produktene, og oppbevare informasjonen om produktene ett ledd tilbake og ett ledd frem i en periode på fem år.
Forordningen fastsetter krav om at opplysninger om godkjente GMO skal registreres i et åpent register, og den hjemler også en rekommandasjon om prøvetaking og analyse.
Bestemmelsene om sporbarhet gjelder ikke for spormengder av GMO i mat og fôr som ikke overstiger de terskelverdier som er satt.
Mer om unike identifikasjonskoder. Sporbarhets- og merkeforordningen er rettslig grunnlag for rettsakten som gir bestemmelser om unike identifikasjonskoder for GMO; forordning (EF) nr. 65/2004 om etablering av et system for utvikling og tildeling av unike koder for genmodifiserte organismer.
Formålet med de unike kodene er å kunne identifisere ulike GMO-er. I et vedlegg til forordningen om unike koder, går det fram hvordan de (alfanumeriske) unike kodene skal utformes. En unik kode kan sies å representere GMO-ens molekylære identitet. Ved å søke opp unike koder, skal man kunne finne opplysninger om de modifiserte egenskapene.
De unike kodene er et sentralt element i systemet for sporbarhet og merking. Alle søknader til EU om godkjenning av en GMO, både etter GM mat- og fôrforordningen og etter utsettingsdirektivet, skal inneholde opplysninger om en unik kode for den omsøkte GMO. Se flere detaljer om bruken av unike koder i Boks 6.1.
Boks 6.1 Unike identifikasjonskoder for genmodifiserte organismer
De fleste GMO har ett eller flere handelsnavn, som benyttes i markedsføring og andre sammenhenger. Dette kan bidra til å skape forvirring om hva det siktes til. For å gjøre det enkelt å sikre at man snakker om den samme GMO-en, har man internasjonalt, gjennom OECD, etablert et system for å gi hver enkelt GMO en såkalt unik identifiseringskode som er en ni tegn lang kode (se tabellen under for eksempler). For eksempel har Monsanto utviklet to typer av soya som er tolerante for herbicidet glyfosat (se tabellen). Disse omtales i mange sammenhenger som RoundupReady soya, og da gjerne som RR1 og RR2 i kortform. Det er den som utvikler og eier rettighetene til den enkelte GMO som melder inn den aktuelle GMO-en i systemet. Det kan skje i forbindelse med søknad om godkjenning (det er blant annet et krav at unik identifiseringskode må være på plass for at en GMO skal kunne bli godkjent i EU), men kan også skje på et tidligere tidspunkt, for eksempel i forbindelse med feltforsøk.
Noen eksempler på GMO-er og deres unike identifikasjonskoder
Art | Kort produktbeskrivelse | Egenskaper endret/tilført | Unik identifikasjonskode |
|---|---|---|---|
Soya | herbicid (glyfosat/Roundup) tolerant (eldste kommersielle variant) | CTP4-epsps tilført | MON-Ø4Ø32-6 |
Soya | herbicid (glyfosat/Roundup) tolerant (andre kommersielle variant) | CTP4-epsps tilført | MON-89788-1 |
Mais | (stacked traits): insektresistent (Bt-toksin) og herbicidtolerant (glufosinat ammonium) | cry1Ab og pat tilført | SYN-BTØ11-1 |
Mais | insektresistent (Bt-toksin) | vip3Aa20 tilført | SYN-IR162-4 |
Mais | insektresistent (Bt-toksin) med seleksjonsmarkør for metabolisme av mannose | mCry3A og pmi tilført | SYN-IR6Ø4-5 |
Mais | herbicidtolerant (glyfosat) | mepsps tilført | MON-ØØØ21-9 |
Mais | (stacked events): krysning av fire av de maisene som er beskrevet over. | se hver enkelt GMO over | SYN-BTØ11-1 × SYN-IR162-4 × SYN-IR6Ø4-5 × MON-ØØØ21-9 |
Mais | (stacked traits): insektresistent (Bt-toksin) og herbicidtolerant (glufosinat ammonium) med antibiotikaresistensgen (ampicillin) som seleksjonsmarkør | cry1Ab, bar og bla tilført | SYN-EV176-9 |
Krysningen av de fire GMO-maisene (eventene) Bt11 (SYN-BTØ11-1), MIR162 (SYN-IR162-4), MIR604 (SYN-IR6Ø4-5) og GA21 (MON-ØØØ21-9) er et eksempel på en stack uten noen egen unik identifikasjonskode. Hver av disse mais-GMO-ene har sin egen unike identifikasjonskode og krysningen identifiseres som SYN-BTØ11-1 × SYN-IR162-4 × SYN-IR6Ø4-5 × MON-ØØØ21-9. Hensikten med å krysse er å oppnå et produkt med et bredere spekter av egenskaper. Krysningen inneholder i dette tilfellet totalt seks ulike egenskaper/traits: en seleksjonsmarkør (pmi) som koder for å bryte ned sukkerarten mannose, to gener som gir toleranse for glufosinat-ammonium og glyfosat herbicider (pat og mepsps), og tre gener som koder for insektresistens (cry1Ab, mcry3A og vip3Aa20) rettet hhv. mot sommerfugler og biller. Slike GMO-er omtales som «stacks», men mer spesifikt er disse stacked events.
Når GMO-er får tilført flere kodende egenskaper, omtales dettes som ‘stacking’. Siden tidlig på 2000-tallet har det i økende grad vært vanlig at GMO-søknader gjelder konvensjonelle krysninger mellom ulike GMO-er (eventer). Slike stackede eventer får ingen unik kode, men koden består i stedet av kombinasjonen av de unike kodene for hver av de enkelte eventene og viser at stacken er en hybrid.
Mange GMO-eventer kan ha fått tilført flere egenskaper uten at dette har medført mer enn én unik kode. Av de syv EU-godkjente enkelt-event soyatypene, er det bare en av dem som har fått tilført kun ett modifisert gen som gir en modifisert egenskap (trait). De øvrige seks har to eller flere tilførte modifiserte gener/traits, og kan benevnes ‘stacked traits’ i stedet for ‘stacked events’.
Flertallet viser til at ikke alle land i verden har krav om at systemet med unike identifikasjonskoder skal benyttes. Land som ikke definerer alle typer av målrettet mutagenese som genmodifisering, vil heller ikke ha krav om at slike organismer skal få en unik identifiseringskode. Eksport av slike vil da kunne skje uten at det medfølger dokumentasjon som forteller en eventuell mottaker i EU/EØS, at produktet er framstilt med genteknologi.
6.4.2.4 Godkjente GMO til mat og fôr i EU
Under forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr er det per april 2023 godkjent cirka 100 ulike varianter GMO av mais, soya, bomull, raps og sukkerbete, og det er cirka 20 søknader under behandling. EU-kommisjonen fører register over alle godkjente GMO under forordningen, se: https://webgate.ec.europa.eu/dyna2/gm-register/.
EU importerer store mengder genmodifisert fôr til produksjonsdyr, og er helt avhengig av denne importen. Det er imidlertid lite genmodifisert mat på markedet.
Kun én GMO er godkjent for dyrking i EU og det er mais Mon 810 (MON-ØØ81Ø-6) som hovedsakelig dyrkes i Spania og litt i Portugal. Maisen dyrkes på cirka 1,5 % av totalt maisareal i EU, tilsvarende cirka 150.000 hektar. Mais Mon 810 er resistent mot larveangrep av sommerfuglen maispyralide på grunn av det innsatte genet cry1Ab fra jordbakterien Bacillus thuringiensis som fører til at maisplantene produserer Cry1Ab proteinet, som er giftig for enkelte insekter. Maispyralide er en betydelig skadegjører i mais i Europa, men i Norge er ikke arten rapportert som skadegjører i jordbruket.
6.4.2.5 Tiltak for å hindre innblanding av GMO i konvensjonelle og økologiske avlinger
Kommisjonsanbefaling 2010/722/EU21 gir retningslinjer for utvikling av nasjonale tiltak for å forhindre utilsiktet innblanding av GMO i avlinger av konvensjonelle og økologiske sorter som dyrkes i nærheten av arealer hvor det dyrkes godkjent GMO, såkalt sameksistenstiltak.
Kommisjonsanbefalingen er hjemlet i utsettingsdirektivet, men er her omtalt sammen med annet matlovsregelverk da det er besluttet at sameksistensregelverk i Norge skal hjemles i matloven og ikke i genteknologiloven (se kapittel 6.4.5.8). Formålet med tiltakene skal være å unngå potensielle økonomiske tap og andre ulemper som følge av innblanding av GMO i ikke-genmodifiserte avlinger, i områder der dyrking av GMO er tillatt.
Kommisjonsanbefalingen er ikke bindende for medlemslandene, men kun retningsgivende for hvilke generelle prinsipper som bør følges ved utvikling av sameksistenstiltak som dyrkingsregler og regler for kompensasjon og erstatning. Det følger av anbefalingen at tiltakene bør være proporsjonale når det gjelder å skulle beskytte konvensjonelle og økologiske avlinger mot innblanding av GMO, og de bør være innrettet slik at ingen av produksjonsformene innebærer unødvendige byrder.
6.4.2.6 Kontrollforordningen
Forordning (EU) nr. 2017/625 om offentlig kontroll22 retter seg hovedsakelig mot offentlige myndigheter, og stiller krav til organisering og gjennomføring av risikobasert kontroll på områdene for næringsmiddel- og fôrvareregelverket samt reglene for dyrs helse og velferd, plantehelse og plantevernmidler, for å sikre etterlevelse av regelverket som gjelder for matkjeden. Forordningen skal sikre et høyt vernenivå for menneskers og dyrs helse og miljøet, og omfanget er nylig utvidet betraktelig og omfatter nå også plantehelse, plantevernmidler, animalske biprodukter, økologiske produkter, GMO på mat- og fôrområdet og beskyttede betegnelser for næringsmidler. Dette sikrer i større grad én rettslig ramme for alle kontrollbestemmelsene i matkjeden. Formålseffektivitet skal oppnås gjennom tydeligere krav til ressurser, opplæring, risikobasering, gjennomføring av kontroll, etatsrevisjon, EU-inspeksjon m.m. Videre stilles det økte krav til at dokumenter og rutiner skal være offentlig tilgjengelige. Når det gjelder kontrollforordningens saklige virkeområde i Norge, vises det til kapittel 6.4.5.5 om kontrollforskriften.
6.4.2.7 Åpenhetsforordningen
Forordning (EU) nr. 2019/1381 om åpenhet og bærekraft i EUs risikovurdering i næringsmiddelkjeden (kortnavn åpenhetsforordningen), har sitt grunnlag i General Food Law og gjennomfører en rekke endringer i denne. Forordningen gjør endringer i EFSAs rutiner for å oppnå bedre kvalitet og styrke påliteligheten av risikovurderingene, øker åpenheten i risikoanalysen, forbedrer risikokommunikasjonen og legger til rette for bedre styring av EFSA. Rettsakten skal bidra til økt transparens og åpenhet gjennom å gi allmennheten større rett til innsyn i data som industrien har sendt til EFSA i forbindelse med risikovurderinger av et produkt, for eksempel en GMO eller et plantevernmiddel. For å møte kritikken om at EFSAs vurderinger kun baseres på industriens egen studier, skal EFSA også gjøre egne søk i vitenskapelig litteratur slik at de kan ta hensyn til andre eksisterende data og studier. I utgangspunktet vil alle data som industrien fremlegger som ledd i en søknadsprosess automatisk offentliggjøres, såfremt det ikke foreligger særlig grunn til noe annet.
Åpenhetsforordningen endrer General Food Law (GFL) og syv rettsakter særlig knyttet til søknader om godkjenninger med hensyn til godkjenningskrav og risikovurderinger for ulike produktgrupper: seks forordninger med hjemmel i GFL herunder forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr, samt utsettingsdirektivet 2001/18/EF.
6.4.3 Matområdet i Norge – generelt og spesielt regelverk for genmodifisert mat og fôr
6.4.3.1 Innledning
Parallelt med EUs matreform på 2000-tallet (se kapittel 6.4.1), vurderte norske myndigheter forenklinger av næringsmiddellovgivningen og en mer effektiv organisering av tilsynsvirksomheten på mattrygghetsområdet. Resultatet var en modernisering av matforvaltningen i 2004 og opprettelsen av Mattilsynet under de tre matdepartementene Helse- og omsorgsdepartementet, Landbruks- og matdepartementet og Nærings- og fiskeridepartementet. Mattilsynet er departementenes utøvende organ på matområdet med ansvar for hele matkjeden og utarbeider forslag til regelverk og har hovedansvaret for tilsyn med at regelverket på matområdet etterleves.
Vitenskapskomitéen for mattrygghet (heretter VKM) ble opprettet i 2004 for å dekke matforvaltningens behov for uavhengige, vitenskapelige risikovurderinger, og er Norges parallell til EUs risikovurderingsorgan EFSA. Senere har også miljømyndighetene tatt i bruk VKM for uavhengige risikovurderinger på flere områder, og VKM endret i 2017 navn til Vitenskapskomitéen for mat og miljø.
Kunnskapsinstitusjonene på matområdet (Veterinærinstituttet, Folkehelseinstituttet, Havforskningsinstituttet og Norsk Institutt for Bioøkonomi (NIBIO)) gir faglige råd og støtte til matdepartementene og Mattilsynet.
Det viktigste målet i norsk matpolitikk er å sikre helsemessig trygg mat til forbrukerne i Norge og i de markeder vi eksporterer til. I tillegg er det et helt sentralt mål at maten oppleves som trygg. I dette ligger det at myndighetene skal ha et spesielt fokus på forbrukernes behov for informasjon og kunnskap.
Lov om matproduksjon og mattrygghet mv. (matloven) etablerer et felles hjemmelsgrunnlag for regulering av forhold langs hele matproduksjonskjeden fra primærproduksjon til omsetning – fra jord/fjord til bord («farm-to-fork»). Loven trådte i kraft i forbindelse med opprettelsen av Mattilsynet i 2004 og samlet helt eller delvis 13 forskjellige lover. Med et slikt spenn i omfanget av forhold som skal reguleres, kan matloven ikke inneholde detaljreguleringer innenfor hvert felt. Det ville blitt uoversiktlig og lite hensiktsmessig. Loven er derfor begrenset til å fastlegge de viktigste prinsippene og pliktene vedrørende produksjon langs hele produksjonskjeden, hovedreglene for den offentlige kontrollen med denne produksjonen, samt hjemler for å ivareta viktige hensyn som ikke direkte retter seg mot matproduksjonen. Matloven er en fullmaktslov, det vil si at den gir utstrakte hjemler til å gi mer detaljerte bestemmelser på forskriftsnivå både for gjennomføring av EØS-relevante rettsakter på matområdet, og rent nasjonale forskrifter. Per i dag hjemles 244 forskrifter i matloven, hvorav flesteparten er gjennomføring av EØS-regelverk.
Oppsummert omfatter matloven og matområdet produksjon, bearbeiding og distribusjon av innsatsvarer og næringsmidler, herunder drikkevann. Loven omfatter også alle forhold i forbindelse med produksjon av materialer og gjenstander som er bestemt til å komme i kontakt med, eller kan ha innvirkning på innsatsvarer eller næringsmidler. Videre omfatter loven all bruk av innsatsvarer, og alle forhold vedrørende plante- og dyrehelse, herunder produkter, gjenstander og organismer som kan føre med seg smitte.
I tråd med sektoransvaret er Mattilsynet ansvarlig myndighet for alle hensyn som skal ivaretas for hele matkjeden, herunder mattrygghet, plante- og dyrehelse, dyrevelferd, miljøhensyn og miljøvennlig produksjon, kvalitet og forbrukerinteresser. Virkeområdet er bredt for å fange opp hele kjeden fra fjord/jord til bord og kunne omfatte alle bestemmelser som har direkte eller indirekte innvirkning på næringsmidler og innsatsvarer. Alle ledd i produksjon, bearbeiding og distribusjon av næringsmidler og innsatsvarer er omfattet, blant annet med godkjenning av genmodifisert, prosessert mat og fôr, plantevernmidler, såvarer og godkjenning av plantesorter.
6.4.3.2 Forpliktelser etter EØS-avtalen
Matområdet (i vid forstand) er en del av EØS-avtalen. Den samme helkjedetankegangen og de faglige prinsippene som ligger til grunn for EUs General Food Law (se kapittel 6.4.1), er tatt hensyn til i matloven og General Food Law er gjennomført i Norge ved en såkalt henvisningsforskrift hjemlet i matloven.
Norge er etter EØS-avtalen forpliktet til å følge EU-reglene som gjelder blant annet for mattrygghet og matproduksjon. Rettsaktene på matområdet utgjør over 40 prosent av rettsaktene i EØS-avtalen, og matområdet er i volum det største rettsområdet i EØS-avtalen23.
EUs lovgivning på mat- og veterinærområdet er i hovedsak innlemmet i EØS-avtalen og gjennomført i norsk rett. Som tidligere nevnt, er derimot EUs omfattende regelverk for genmodifisert mat og fôr (12 rettsakter), den såkalte GM-pakken, ennå ikke innlemmet i EØS-avtalen. Utsettingsdirektivet er en del av EUs miljøregelverk og er gjennomført i genteknologiloven (se kapittel 6.2).
6.4.3.3 Regelverksutvikling genmodifisert mat og fôr – historikk
Forvaltningen av regelverk om genmodifiserte produkter er i Norge delt mellom mat- og helseforvaltningen og miljøforvaltningen. Matforvaltningen er godkjenningsmyndighet for genmodifisert mat og fôr fra levende GMO, det vil si prosesserte/bearbeidede produkter som er antatt ikke-reproduserbare. Forskrifter under matloven regulerer likevel merking både av levende og bearbeidet GMO til mat og fôr, merking av genmodifiserte plantesorter og visse typer forbud mot levende GMO. For genmodifiserte mat- og fôrplanter godkjent for dyrking i henhold til genteknologiloven, kan det ikke omsettes såvarer av disse før plantesorter utviklet på basis av de godkjente GMO-ene står på EUs felles sortsliste, eller er godkjent i henhold til forskrift om prøving og godkjenning av plantesorter hjemlet i matloven (se kap. 6.4.5.1.6). For at en sort skal oppføres på EUs felles sortsliste, må sorten først være søkt og godkjent for oppføring på minimum et EU- eller EØS-lands nasjonale sortsliste.
I 1997 ble det fastsatt krav om merking av genmodifisert mat, men tilsetnings- og aromastoffer laget fra GMO var unntatt fra kravet. Et drøyt år etterpå kom det godkjenningskrav for genmodifisert mat, men tilsetnings- og aromastoffer fra GMO var også unntatt dette kravet. I oktober 1999 ble det innført krav om merking av fôrvarer, men kun for produkter hvor DNA eller protein som stammer fra genmodifiseringen kunne påvises. Godkjenning av levende GMO til bruk som fôr var regulert av genteknologiloven fra 1. september 1993, mens godkjenningskrav for prosessert genmodifisert fôr ble innført høsten 2005.
I juni 1997 fattet Stortinget vedtak om å «forby produksjon, import og omsetting av alle genmanipulerte produkter som inneholder gener som koder for antibiotikaresistens, og å arbeide for internasjonale forbud på dette området.» Daværende Statens næringsmiddeltilsyn (SNT), Statens landbrukstilsyn og Fiskeridirektoratet implementerte forbudet med egne forskrifter, men disse trådte ikke i kraft før det var utviklet analysemetoder for å påvise funksjonelle antibiotikaresistensgener tilført ved genmodifisering. Veterinærinstituttet utviklet disse metodene på oppdrag fra SNT.
Da EU vedtok nytt regelverk for genmodifisert mat og fôr i 2003 («one door – one key principle», se kapittel 6.4.2.1), endret Mattilsynet på oppdrag fra matdepartementene, nasjonalt regelverk under matloven i påvente av at EU-regelverket skulle innlemmes i EØS-avtalen. Det reviderte norske regelverket inneholder de viktigste elementer fra EUs regelverk med godkjennings- og merkekrav for genmodifiserte produkter, og trådte i kraft høsten 2005. Regelverket er ikke en formell eller fullstendig gjennomføring av EUs forordninger på området.
EUs regelverk (den såkalte GM-pakken) er fortsatt ikke innlemmet i EØS-avtalen, og genmodifisert mat og fôr som er godkjent i EU, er ikke lovlig å omsette i Norge. Virksomheter som ønsker å selge genmodifisert mat og fôr i Norge, må derfor sende separate søknader til Norge. Våren 2023 behandler Mattilsynet den første søknaden til Norge om godkjenning av en rapsolje med øket innhold av langkjedete omega-3 fettsyrer fra genmodifisert raps, til bruk som ingrediens i fiskefôr24.
Figur 6.2 viser skjematisk og overordnet den historiske utviklingen av krav i nasjonalt regelverk for genmodifiserte produkter (unntatt legemidler). Utviklingen viser at det har blitt godkjennings- og merkekrav for stadig flere produktgrupper.
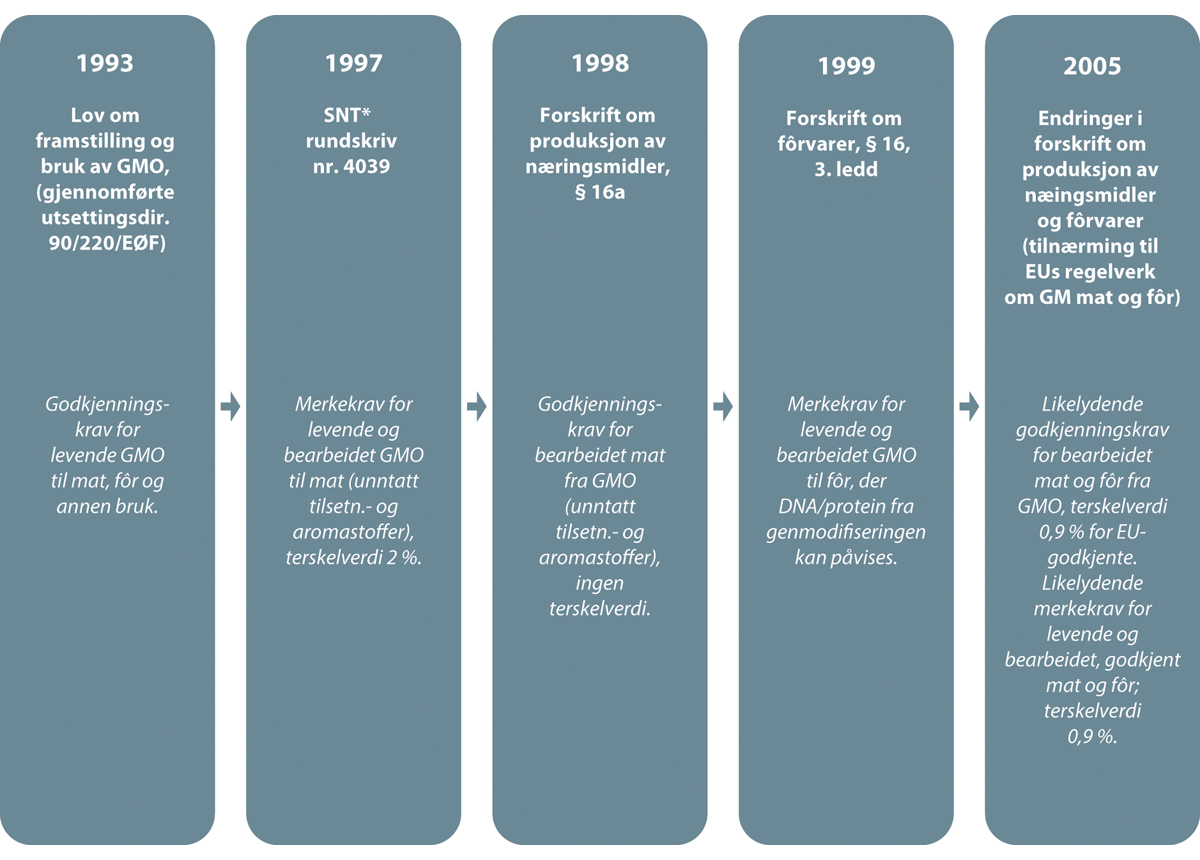
Figur 6.2 Oversikt over utviklingen av regelverk for genmodifiserte produkter (unntatt legemidler) i Norge med hensyn til krav om godkjenning og merking. Regelverkene er både EØS-regelverk og rent nasjonalt regelverk.
* Statens næringsmiddeltilsyn; gikk inn sammen med andre etater i det nyopprettede Mattilsynet i 2004.
6.4.4 Bestemmelser i matloven som hjemler forskrifter om genmodifiserte produkter
Matloven hjemler bestemmelser om godkjenningskrav for prosessert, genmodifisert mat og fôr, og merkekrav for både levende og prosessert GMO til mat og fôr. Kravene er i hovedsak de samme som EUs regelverk på området.
6.4.4.1 Matloven kapittel I: Formål, virkeområde og definisjoner
Matloven skal ivareta et helkjedeperspektiv og en helhetlig forvaltning fra jord/fjord til bord. Lovens formål fremgår av § 1 og lyder:
«Formålet med loven er å sikre helsemessig trygge næringsmidler og å fremme helse, kvalitet og forbrukerhensyn langs hele produksjonskjeden, samt ivareta miljøvennlig produksjon.
Loven skal videre fremme god plante- og dyrehelse.
Loven skal også ivareta hensynet til aktørene langs hele produksjonskjeden, herunder markedsadgang i utlandet.»
Virkeområdet er bredt for å fange opp hele kjeden fra fjord/jord til bord og kunne omfatte alle forhold som har direkte eller indirekte innvirkning på næringsmidler og innsatsvarer. Det er viktig å merke seg at virkeområdet omfatter alle ledd i produksjon, bearbeiding og distribusjon av næringsmidler og innsatsvarer.
Lovens saklige virkeområde gjelder i henhold til § 2:
«… alle forhold i forbindelse med produksjon, bearbeiding og distribusjon av innsatsvarer og næringsmidler, herunder drikkevann. Loven omfatter også alle forhold i forbindelse med produksjon av materialer og gjenstander som er bestemt til å komme i kontakt med, eller kan ha innvirkning på innsatsvarer eller næringsmidler. Videre omfatter loven all bruk av innsatsvarer.
Loven omfatter alle forhold vedrørende plante- og dyrehelse, herunder produkter, gjenstander og organismer som kan føre med seg smitte.»
En ‘genmodifisert organisme’ (GMO) og ‘genteknologi’ er definert i genteknologiloven og gjelder også for forskrifter under matloven.
6.4.4.2 Matloven kapittel II: Generelle krav og forpliktelser
Bestemmelsene i matloven gjelder både for konvensjonelle og genmodifiserte innsatsvarer og næringsmidler. Loven gir hjemmel for forskrifter om blant annet produktgodkjenning og begrensning/forbud (§ 9), merking (§ 10) og sporbarhet (§ 11).
Godkjenningskrav
Ordlyden i virkeområdebestemmelsen § 2 («alle forhold») og i § 9 første ledd om produktgodkjenning, innhold, sammensetning og kvalitet, tilsier at matloven gir hjemmel for forskriftsbestemmelser om godkjenning av både levende GMO og bearbeidede GMO produkter til mat og fôr:
«Kongen kan i forskrifter stille krav til innsatsvarer, planter, dyr, næringsmidler, animalske biprodukter samt materialer og gjenstander som er bestemt til å komme i kontakt med eller kan ha innvirkning på innsatsvarer eller næringsmidler, herunder om godkjenning, innhold og kvalitet mv.»
Matloven må likevel tolkes innskrenkende ved en eventuell motstrid. Dette fordi genteknologiloven anses som et mer spesialisert regelverk utviklet for dette særlige området. Genteknologiloven anses dermed som et unntak fra den mer generelle matloven, jamfør lex specialis-prinsippet. Det følger utvetydig av bestemmelsene i genteknologiloven at denne loven gjelder godkjenning av levende GMO.
Godkjenningskrav for prosessert genmodifisert mat og fôr og for visse grupper av plantesorter gis i henholdsvis generell forskrift for næringsmidler, fôrvareforskriften og forskrift om prøving og godkjenning av plantesorter, se omtale lenger ned.
Det fremkommer uttrykkelig i § 9 andre ledd at bestemmelsen om begrensninger og forbud i matloven § 9 både gjelder levende GMO og produkter produsert fra GMO:
«Kongen kan gi forskrifter om begrensning av eller totalforbud mot innhold av genmodifiserte organismer, mot innhold av gener fra genmodifiserte organismer, og produkter fra visse genmodifiserte organismer.»
Dette leddet hjemler det særnorske forbudet mot mat og fôr som inneholder markørgener som koder for antibiotikaresistens. Forbudsbestemmelsene ble gitt etter Stortingsvedtak i 1999 om å forby slike produkter ut fra et så høyt beskyttelsesnivå at det ble vanskelig å innføre totalforbud ut fra vanlig risikotankegang. Forarbeidene til loven nevner at fordi totalforbud er et meget sterkt virkemiddel og man lett kan komme i konflikt med WTO/EØS-avtalene, er det viktig å være varsom med å innføre totalforbud25.
Forbudene mot mat og fôr med innhold av funksjonelle antibiotikaresistensgener tilført ved genmodifisering, gis i fôrvareforskriften § 5 og i en egen forskrift for slike næringsmidler, se omtale lenger ned.
Merkekrav
Hovedhensikten med paragraf 10 om merking, presentasjon og reklame, er at forbrukerne ikke skal villedes og at skal ha mulighet til å foreta informerte valg, jamfør ordlyden i første ledd:
«Virksomheten skal sørge for at merking, presentasjon, reklame og markedsføring er korrekt, gir mottaker tilstrekkelig informasjon og ikke er egnet til å villede.»
Dette er også nedfelt i Food Law (gjennomført i matlovsforskriften), hvor det fremgår at regelverket
«… skal hasom mål å verne forbrukerinteressene og gi forbrukerne et grunnlag for å foreta velbegrunnede valg med hensyn til de næringsmidler de konsumerer. Det skal ha som mål å forebygge; a) bedragersk eller villedende praksis, b) forfalskning av næringsmidler og c) enhver annen praksis som kan villede forbrukeren.»
Andre ledd i § 10 gir hjemmel for å
«gi nærmere forskrifter om merking, presentasjon og reklame, herunder om forbud mot helsemessig uønsket markedsføring og om vilkår for bruk av frivillige merkeordninger.»
Bestemmelsen om merking i matloven § 10 gir hjemler til å i forskrift gi bestemmelser om merking av både levende og prosessert/bearbeidet GMO til bruk som mat, fôr eller såvare. Forskrift om merking mv. av GMO § 2, som er hjemlet i genteknologiloven, unntar GMO mat, fôr og såvarer fra merkekrav etter genteknologiloven, jamfør andre ledd siste setning: «Næringsmidler, fôrvarer og såvarer er unntatt fra bestemmelsene i § 19, første og annet ledd». Levende GMO godkjent etter genteknologiloven som skal brukes som mat, fôr eller såvare, skal etter dette merkes i henhold til krav gitt med hjemmel i matloven. Enkelte andre paragrafer i matloven gir også bestemmelser om levende GMO og omtales nedenfor.
Merkekrav for levende og prosessert genmodifisert mat, fôr og for såvarer gis i hhv. generell forskrift for næringsmidler, fôrvareforskriften og forskrift om såvarer, se omtale lenger ned.
Sporbarhetskrav
Matloven § 11 om sporbarhet åpner for at det kan gis forskrifter om sporbarhet:
«Kongen kan gi forskrifter om sporbarhet for innsatsvarer, planter, dyr eller næringsmidler, samt om sporbarhet for materialer og gjenstander som er bestemt til å komme i kontakt med, eller kan ha innvirkning på innsatsvarer eller næringsmidler.»
Denne bestemmelsen gir rettslig grunnlag for at matlovsforskriften har gjennomført General Food Law og herunder dens artikkel 18 om sporbarhet.
Fastsettelse av sporbarhetskrav omhandler gjerne sporing i alle ledd i produksjon, bearbeiding og distribusjon. I forarbeidene til matloven (Ot.prp. nr. 100 (2002–2003)) omtales sporbarhetskravet som etablering av systemer hos fôr- eller næringsmiddelvirksomheter som gjør dem i stand til å identifisere hvem de har mottatt fôr, næringsmidler eller dyr som brukes i matproduksjon fra, og hvem de har levert slike til (Ot.prp. kapittel 8.6.1).
Sporbarhet gjør det enklere å trekke næringsmidler tilbake fra markedet, samtidig med at forbrukerne kan gis mer målrettede og presise opplysninger om produktene (Ot.prp. kapittel 8.6.2). Sporbarhetskravet ivaretar også forhold hvor det ikke er helsefare forbundet med videre omsetning, men hvor forbrukere erfarer at et produkt ikke svarer til de forventninger de har hatt. I slike tilfeller er det rimelig at det er den aktuelle produsent eller leverandør som blir markedsmessig ansvarlig – og ikke alle leverandører som produserer den aktuelle vare – slik mønsteret vil være ved generisk markedsføring (Ot.prp. kapittel 8.6.2).
Sporbarhetskrav kan være både generelle og spesielle. Det er ikke laget eksplisitte krav til sporbarhet for godkjente genmodifiserte produkter, men det kan stilles vilkår om sporbarhet i forbindelse med godkjenning av genmodifisert mat og fôr. Dette omtales lenger ned.
6.4.5 Forskriftsbestemmelser om genmodifiserte produkter
De norske forskriftsbestemmelsene med krav til genmodifisert mat og fôr inneholder de samme elementene som rettsaktene i GM-pakken som ennå ikke er innlemmet i EØS-avtalen.
6.4.5.1 Godkjenningskrav og unntak
6.4.5.1.1 Godkjenningskrav genmodifisert mat og fôr
Generell forskrift for næringsmidler § 16a inneholder godkjenningskrav til alle typer av prosesserte genmodifiserte næringsmidler, jamfør første ledd:
«Næringsmidler og næringsmiddelingredienser herunder tilsetningsstoffer og aromastoffer som består av, inneholder eller er fremstilt på grunnlag av genmodifiserte organismer, er ikke tillatt omsatt eller markedsført med mindre Mattilsynet har godkjent dette.»
Tilsvarende krav gjelder for fôrvarer, jamfør forskrift om fôrvarer § 4a første ledd:
«Fôrvarer herunder tilsetningsstoffer som består av, inneholder eller er fremstilt på grunnlag av genmodifiserte organismer, er ikke tillatt omsatt eller markedsført med mindre Mattilsynet har godkjent dette.»
Vedtak om godkjenning eller avslag etter begge bestemmelsene fattes av Mattilsynet hovedkontoret, og Landbruks- og matdepartementet er klageinstans.
6.4.5.1.2 Unntak fra godkjenningskravet
Unntatt fra kravet om godkjenning etter første ledd i begge forskrifter, gjelder for
«produkter som omfattes av lov 2. april 1993 nr. 38 (genteknologiloven)»
Det er videre unntak fra kravet om godkjenning for
«produkter som er fremstilt ved hjelp av genmodifiserte organismer, men hvor sluttproduktet ikke inneholder materiale fra det opprinnelige genmodifiserte materiale, herunder genmodifiserte…»
(mat) «… prosesshjelpemidler og næringsmidler som er behandlet med genmodifiserte prosesshjelpemidler»
(fôr) «… genmodifiserte prosesshjelpemidler samt fôrmidler og tilsetningsstoffer som er behandlet med genmodifiserte prosesshjelpemidler»
Produkter fremstilt ved hjelp av genmodifiserte organismer er for eksempel animalske produkter produsert fra dyr fôret med genmodifisert fôr (kjøtt- og fiskeprodukter). Fermenteringsprodukter er en annen gruppe produkter som er fremstilt ved hjelp av genmodifiserte mikroorganismer i lukkede systemer. Produktene kan være enkelte vitaminer, aminosyrer og enzymer. Tekniske hjelpestoffer er heller ikke omfattet av godkjennings- og merkekravene.
Godkjenningsplikten gjelder heller ikke for sporforurensninger, jamfør andre ledd nr. 1 og 2 som er likelydende i generell forskrift og fôrforskriften, og som lyder:
«Godkjenningsplikt etter første ledd gjelder ikke ved utilsiktet eller teknisk uunngåelig tilstedeværelse av genmodifisert materiale forutsatt at virksomheten kan dokumentere dette, samt forutsatt:
1. tilstedeværelse opp til 0,9% dersom det genmodifiserte materialet er godkjent i EU,
eller
2. tilstedeværelse opp til 0,5% dersom det genmodifiserte materialet har vært risikovurdert og er funnet helsemessig trygt av enten EFSA/EUs vitenskapskomiteer eller den norske Vitenskapskomiteen for Mattrygghet samt at analysemetodikk er offentlig tilgjengelig.»
Unntaket for sporforurensning av EU-godkjent materiale er en særnorsk regel. EU-godkjente GMO er vurdert av EFSA å være helsemessig trygge, og Norge aksepterer derfor slike små forurensninger. Bakgrunnen for terskelverdien, er at det kan være vanskelig for importørene og industrien å oppnå garantier fra leverandører for 100 prosent fravær av genmodifisert materiale i råstoff og bearbeidede produkter.
For begge unntakene gjelder at terskelverdiene relaterer seg til ingrediensnivå, for eksempel vil andelen genmodifisert soya i et sammensatt produkt regnes ut som andel av soyaingrediensen i produktet.
Unntakene fra godkjenningskravet for slike sporforurensninger av visse typer av genmodifisert materiale, forutsetter at virksomheten kan dokumentere at forurensningen er utilsiktet eller teknisk uunngåelig, og at det er iverksatt tiltak for å unngå forurensning med slikt materiale. Terskelverdiene er maksimumsverdier, og det er virksomheten selv som må dokumentere at innholdet av genmodifisert materiale er under terskelverdiene, at innholdet er utilsiktet eller teknisk uunngåelig, og at det er godkjent i EU. Aktørene må derfor arbeide for å finne kildene til eventuelle spormengder i varepartiene og tilstrebe en så lav sporforurensing som mulig.
Både i Norge og i EU er det en absolutt nulltoleranse for ikke EU-godkjente GMO, det vil si at i andre tilfeller enn de unntakene som er beskrevet ovenfor, er godkjenningsplikten absolutt.
6.4.5.1.3 Analyser, prøvemateriale, sporbarhet og unik identifikasjonskode
Det følger av tredje og fjerde ledd i henholdsvis § 16a og § 4a i de to forskriftene at
«I forbindelse med søknad eller godkjenning kan Mattilsynet stille krav om analysemetodikk, prøvemateriale og sporbarhet mv.» og at
«Det stilles krav om angivelse av unik identifikasjonskode for genmodifiserte organismer i ledsagende dokument for produkter som består av eller inneholder slike organismer i forskrift 2. september 2005 nr. 1009 om merking, transport, import og eksport av genmodifiserte organismer.»
Å ha tilgang til spesifikke analyser og prøvemateriale for påvisning av GMO er viktig for tilsynsmyndigheten i forbindelse med offentlig kontroll. Derfor kan det settes krav om dette ved nasjonale søknader på samme måte som for GMO som godkjennes i EU under forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr. Slike, og andre krav kan også forskriftsfestes, jamfør det sjette leddet:
«Mattilsynet kan fastsette utfyllende forskrifter for gjennomføringen av bestemmelsene i denne paragraf.»
For GMO som godkjennes i EU til mat og fôr, har Norge på lik linje med medlemsstatene tilgang til analysemetoder for påvisning av de godkjente GMO-ene.
6.4.5.1.4 Mer om sporbarhetskrav
I de samme to godkjenningsparagrafene fremgår det at i forbindelse med søknad eller godkjenning, kan Mattilsynet stille krav om analysemetodikk, prøvemateriale og sporbarhet mv.
Å ha tilgang til spesifikke analyser og prøvemateriale for påvisning av GMO, er viktig for tilsynsmyndigheten i forbindelse med offentlig kontroll. Derfor kan det settes krav om dette ved nasjonale søknader på samme måte som for GMO som godkjennes i EU under forordning (EF) 1829/2003 om genmodifisert mat og fôr.
Sporbarhetskrav må ikke forveksles med dokumentasjonskontroll knyttet til utilsiktet eller teknisk uunngåelig tilstedeværelse av genmodifisert materiale. Det finnes generelle sporbarhetskrav som gjelder for alle næringsmidler og fôrvarer, inkludert de genmodifiserte. Virksomhetene må ha systemer og prosedyrer for å identifisere hvem produktene kommer fra og hvem de videresendes til, og informasjonen om dette må oppbevares i fem år. Spesifikke sporbarhetskrav for genmodifiserte produkter har som hensikt å tilrettelegge for korrekt merking, helse- og miljømessig overvåkning og eventuelt tilbakekalling av produkter. Muligheten for at Mattilsynet kan stille krav om spesifikk sporbarhet i forbindelse med søknad eller godkjenning, ble valgt for å gjennomføre nasjonale regler for sporbarhet som ligger tett opptil EUs regler ved en eventuell godkjenning etter nasjonalt regelverk under matloven. EU krever at den som omsetter genmodifiserte næringsmidler eller fôrvarer skal videreformidle til neste ledd i kjeden informasjon om hver ingrediens som er produsert fra GMO, eller at produktet er produsert fra GMO dersom det ikke finnes noen ingrediensliste.
Det er unntak fra sporbarhetskravet for emballerte produkter som allerede er omfattet av andre identifikasjonssystemer som lot- og batchnummer, forutsatt at denne informasjon fremgår av emballasjen og at informasjonen beholdes i fem år. Virksomheter som omsetter til sluttbruker, som restaurantgjester og vanlige forbrukere, er ikke pliktige til å vite hvem varen er solgt til. Det er også unntak fra kravet om sporbarhet for materiale som skal brukes direkte som næringsmidler/fôrvarer eller til videreforedling og som er utilsiktet til stede i et produkt. Terskelverdien for utløsing av sporbarhetskravene i EU er 0,9 % på ingrediensnivå.
Når det gjelder generelle sporbarhetskrav, gjelder de samme reglene i Norge som i EU. Det følger av Food Law artikkel 18 om sporbarhet at:
1. Næringsmidler, fôr, dyr bestemt til næringsmiddelproduksjon og alle andre stoffer som er bestemt til eller kan forventes å bli iblandet et næringsmiddel eller et fôr, skal kunne spores i alle ledd i produksjon, bearbeiding og distribusjon.
2. Driftsansvarlige for næringsmiddel- og fôrforetak skal kunne identifisere enhver person som har levert til dem et næringsmiddel, fôr, dyr bestemt til næringsmiddelproduksjon eller ethvert stoff som er bestemt til eller kan forventes å bli iblandet et næringsmiddel eller et fôr. For dette formålet skal de driftsansvarlige ha ordninger og framgangsmåter for å kunne gjøre disse opplysningene tilgjengelige for vedkommende myndigheter på anmodning.
De generelle sporbarhetskravene vil også gjelde for levende GMO godkjent etter genteknologiloven, så lenge det er produkter som også er regulert av matloven og matlovsforskriften. Dette vil for eksempel gjelde prydblomster og akvariefisk, i tillegg til mat og fôr. Se mer om sporbarhet i kapittel 6.5.
De generelle sporbarhetskravene er viktige for å raskt kunne fjerne utrygge produkter fra markedet hvis et ledd i kjeden, eller myndigheten via ulike systemer, får informasjon om at et produkt kan være utrygt. Det påligger virksomhetene et stort ansvar i å ha systemer og rutiner for sporbarhet og hvordan, og i hvilke situasjoner man trekker tilbake et produkt (evt. tilbakekaller hvis produktet har nådd forbrukeren). Virksomhetene er også pålagt å varsle tilsynsmyndigheten og kunder i de tilfeller det er aktuelt.
I andre tilfeller er det myndigheten som blir varslet av EUs felles varslingssystem RASFF (se kapittel 6.4.1 og fotnote 14) eller på annen måte fra andre myndigheter, og setter i gang sporing sammen med aktuelle virksomheter.
For å sikre målrettet tilbaketrekking av mat og drikke, må produktene kunne spores. Et spesifikt produkt må kunne spores ett ledd fram (mottaker) og ett ledd tilbake (leverandør). Produktene kan ofte identifiseres via lotnummer, produksjonsdato og lignende. Oppbevaring av faktura og skriftlige registreringer i mottakskontroll kan være viktig dokumentasjon på sporbarhet.
Sporbarhet og systemer for å håndtere sporbarhetsopplysninger gir ikke trygge næringsmidler i seg selv, men er en forutsetning for at man skal kunne trekke tilbake næringsmidler som ikke er trygge.
6.4.5.1.5 Overgangsordning for produkter det ikke tidligere var godkjenningskrav for
Det femte leddet i henholdsvis § 16 a og § 4a innførte en overgangsordning for produktgrupper det ikke var godkjenningskrav til før 15. september 2005:
«Tilsetningsstoffer og aromastoffer som skal godkjennes etter første ledd, og som allerede finnes på det norske markedet pr. 15. september 2005, kan fortsatt omsettes forutsatt at det innen 6 md. etter 15. september 2005 er sendt melding til Mattilsynet med informasjon om stoffene. Innen tre år etter 15. september 2005 må det for disse stoffene søkes om godkjenning etter § 16a.» (mat)
«Fôrvarer og tilsetningsstoff som allerede finnes på det norske markedet pr. 15. september 2005, kan fortsatt omsettes forutsatt at det innen 15. mars 2006 er sendt melding til Mattilsynet med informasjon om fôrvaren eller tilsetningsstoffet. Innen 15. september 2008 må det for disse fôrvarene og tilsetningsstoffene søkes om godkjenning etter § 4a.» (fôr)
Bakgrunnen for overgangsordningen var å gi virksomheter som brukte produkter det ikke tidligere var knyttet godkjenningskrav til, tid til å endre importrutiner eller eventuelt å forberede søknader om godkjenning. Før 15. september 2005 var det ikke godkjenningskrav for prosesserte, genmodifiserte tilsetningsstoffer og aromastoffer til bruk i næringsmidler og for fôrtilsetningsstoffer og fôrvarer i Norge. Slike produkter var da tillatt å omsette så lenge produktene ble merket korrekt. Det er normal prosedyre å fastsette ulike typer overgangsordninger i regelverk slik at virksomheter får anledning til å tilpasse seg det nye regelverket.
Det fulgte av regelverket at virksomheter som meldte inn aktuelle EU-godkjente produkter innen oppgitt frist, kunne fortsette å bruke disse i tre år. Skulle de fortsatt brukes etter dette, måtte virksomhetene søke om godkjenning etter det nye regelverket. Nitten ulike EU-godkjente genmodifiserte produktgrupper ble omfattet av overgangsordningen etter at interesseorganisasjonen Fiskeri- og havbruksnæringens landsforening (heretter FHL; i dag heter foreningen Sjømat Norge) meldte inn disse på vegne av fire fiskefôrvirksomheter i mars 2006. Disse hadde det tidligere ikke vært forbudt å omsette som fiskefôr på det norske markedet.
Da overgangsordningen gikk ut og EUs regelverk om genmodifisert mat og fôr ennå ikke var tatt inn i norsk rett, søkte FHL høsten 2008 om dispensasjon fra å søke godkjenning for de samme genmodifiserte produktene som var omfattet av overgangsordningen. FHL begrunnet søknaden om dispensasjon i at de fire fôrvirksomhetene ønsket å ha muligheten åpen dersom fôrtilgangen skulle tilsi at det ble nødvendig å bruke genmodifisert fôringredienser, at de ønsket konkurranselikhet innenfor EØS-området for valg av fiskefôr og at de ønsket å ha anledning til å kunne bruke disse i egne fôrutviklingsprosjekter.
Dispensasjonen ble innvilget av Mattilsynet i samråd med daværende Fiskeri- og kystdepartementet (FKD) og Landbruks- og matdepartementet (LMD) for ett år. FHL søkte senere om forlengelse av dispensasjonen flere ganger, og Mattilsynet innvilget dispensasjon for ett år av gangen under gitte vilkår etter føringene gitt av FKD og LMD i 2008.
Etter å ha innvilget dispensasjon flere år på rad, foretok Mattilsynet en helhetlig gjennomgang av dispensasjonsmuligheten for FHL. Bakgrunnen for de nye nasjonale godkjenningskravene for genmodifisert mat og fôr med hjemmel i matloven som trådte i kraft i 2005, var at Norge skulle ha et regelverk som var like omfattende som EUs nye regelverk i perioden frem til EUs regelverk var innlemmet i EØS-avtalen og gjennomført i norsk lov. I EU ble stadig flere GMO godkjent med basis i EFSAs helse- og miljørisikovurderinger, mens EFTA-prosessen med innlemming av regelverket ikke skjedde. Dette var på den ene siden et argument for å opprettholde muligheten for å innvilge dispensasjon fra godkjenningskravet. På den annen side hadde ikke de fire angjeldende fiskefôrvirksomhetene benyttet seg av anledningen til å kunne bruke genmodifiserte fôrmidler. Mattilsynet mente derfor at det faktiske behovet ikke lenger forelå. Det er et krav om at det må foreligge særskilte grunner for å innvilge en dispensasjon fra godkjenningskravet for genmodifisert fôr. Når dette ikke lenger forelå, avslo derfor Mattilsynet i 2014 den siste søknaden fra FHL om dispensasjon fra godkjenningskravet for fire fiskefôrvirksomheter.
6.4.5.1.6 Godkjenningskrav for GMO plantesorter og såvarer
For å kunne omsette såvarer av de mat- og fôrvekstene som er vurdert å være de viktigste i EU og Norge, er det krav om at plantesorten må være godkjent og stå oppført på norsk offisiell sortsliste eller EUs felles sortsliste. Disse bestemmelsene er fastsatt i forskrift om prøving og godkjenning av plantesorter. Et vilkår for å bli godkjent er at sorten er skillbar fra andre sorter, ensartet og stabil over tid. Videre er det for opptak på norsk offisiell sortsliste, krav om at kornsorter og sorter av fôrvekster, skal testes ut i felt ulike steder i landet. Dette gjøres sammen med allerede godkjente sorter, for å se om de er like gode eller bedre enn disse i en eller flere egenskaper. Kravene gjelder også genmodifiserte sorter av de aktuelle artene, selv om den omsøkte sorten allerede er godkjent med hjemmel i genteknologiloven.
6.4.5.2 Forbud mot produkter med antibiotikaresistensgener
Forskrift om forbud mot visse genmodifiserte næringsmidler og næringsmiddelingredienser og § 5 i forskrift om fôrvarer, forbyr begge genmodifisert mat og fôr med innhold av funksjonelle antibiotikaresistensgener der disse er tilført ved genmodifisering og kan påvises i sluttproduktet. Forbudene omfatter både GMO omfattet av genteknologiloven, inkludert direktivgodkjente GMO, og produkter omfattet av matloven.
Mat: «Det er forbudt å produsere, importere og omsette næringsmidler og næringsmiddelingredienser som…
Fôr: «Det er forbudt å tilvirke, importere og framby fôrvarer som…
(felles) «inneholder gener som koder for antibiotikaresistens der disse genene er tilført ved genmodifisering og kan påvises i sluttproduktet.»
Dette er et særnorsk regelverk, som er basert på et Stortingsvedtak om særskilt høyt beskyttelsesnivå (se omtale i kapittel 6.4.4.2). I dag er imidlertid bruken av antibiotikaresistensgener som markørgener utfaset, men det er per april 2023 godkjent noen GMO bomull i EU med markørgener som koder for antibiotikaresistens.
6.4.5.3 Merkekrav og unntak
Matinformasjonsforskriften § 4 og forskrift om fôrvarer § 4b gjelder for godkjente genmodifiserte mat- og fôrvarer, og både levende GMO godkjent etter genteknologiloven og bearbeidete/prosesserte produkter godkjent etter matloven omfattes av kravene.
Mat: «Genmodifiserte næringsmidler, herunder tilsetningsstoffer og aromastoffer, skal i varebetegnelsen eller i tilknytning til den aktuelle ingrediensen, merkes med enten «genmodifisert» eller «produsert fra genmodifisert [navn på organismen]» når
1. næringsmidlet består av eller inneholder genmodifiserte organismer, eller
2. næringsmidlet er produsert fra, men ikke inneholder genmodifiserte organismer. Merkeplikten gjelder også for genmodifiserte næringsmidler, tilsetningsstoffer og aromastoffer hvor DNA og protein som stammer fra genmodifiseringen, ikke kan påvises».
Fôr: «Et fôrmiddel eller tilsetningsstoff skal både ved omsetning og når det inngår i en blanding merkes med enten «genmodifisert» eller «produsert fra genmodifisert [navn på organismen]», umiddelbart etter fôrmiddelets eller tilsetningsstoffets spesifikke navn når
1. fôrmiddelet eller tilsetningsstoffet består av eller inneholder genmodifiserte organismer, eller
2. fôrmiddelet eller tilsetningsstoffet er produsert fra, men ikke inneholder genmodifiserte organismer. Merkeplikten gjelder også for genmodifiserte fôrmidler og tilsetningsstoffer hvor DNA og protein som stammer fra genmodifiseringen, ikke kan påvises.»
Genmodifiserte næringsmidler som ikke er ferdigpakket, skal ledsages av de samme opplysningene.
Det er unntak fra merkeplikten for sporforurensninger av godkjent genmodifisert materiale
Mat:
1. «ved utilsiktet eller teknisk uunngåelig tilstedeværelse av genmodifisert materiale i et produkt under 0,9 prosent opp til 0,9 %,» og for
2. «produkter som er fremstilt ved hjelp av genmodifiserte organismer, men hvor sluttproduktet ikke inneholder materiale fra det opprinnelige genmodifiserte materialet, herunder genmodifiserte tekniske hjelpestoffer og næringsmidler som er behandlet med genmodifiserte tekniske hjelpestoffer».
Fôr:
1. «ved utilsiktet eller teknisk uunngåelig tilstedeværelse av genmodifisert materiale i et produkt under 0,9 prosent/i fôrvarer opp til 0,9 %,» og for
2. «produkter som er fremstilt ved hjelp av genmodifiserte organismer, men hvor sluttproduktet ikke inneholder materiale fra det opprinnelige genmodifiserte materialet, herunder genmodifiserte prosesshjelpemidler samt fôrmidler og tilsetningsstoffer som er behandlet med genmodifiserte prosesshjelpemidler.»
Et viktig prinsipp i matforvaltningen er at norske forbrukere skal vite hva maten de spiser inneholder. All mat som omsettes skal være trygg, og forbrukermerking omhandler andre aspekter for eksempel sunnhetsmerking (nøkkelhull), opprinnelsesmerking, eller produksjonsmetode. På den måten settes norske forbrukere i stand til å ta informerte valg, og det sikres et godt forbrukervern.
Alle genmodifiserte mat- og fôrvarer må merkes etter de særskilte kravene. Men siden det per dato ikke er godkjent genmodifisert mat og fôr i Norge, skal det ikke finnes produkter på markedet som er merket ‘genmodifisert’. Av samme årsak skal det ikke være nødvendig å merke produkter med at de ikke inneholder genmodifisert materiale. Dette følger av mer generelle merkekrav som virksomhetene må forholde seg til for å kunne omsette mat og fôr, blant annet matinformasjonsforskriften26 og forskrift om merking mm av fôrvarer27, som begge er implementert EU-regelverk.
Av de generelle kravene er det viktig å fremheve kravet om at merkingen ikke skal være villedende og at man må være forsiktig med såkalt negativ påstandsmerking. En påstand som objektivt sett er sann, kan likevel i enkelte tilfeller eller sammenhenger fremstå som villedende. For eksempel vil appelsiner merket med at de ikke er genmodifiserte ikke kunne godtas av Mattilsynet, fordi ingen appelsiner på markedet er genmodifiserte. Andre negative deklarasjoner som for eksempel ‘100 % fri for GMO’ på et produkt som inneholder soya, mais, eller raps, vil heller ikke godtas da det i de fleste tilfeller i praksis vil være umulig å dokumentere at det ikke er noen sporforurensinger. I andre tilfeller er merkingen kanskje ikke ulovlig, men ikke anbefalt fra myndighetenes side. Det kan finnes varianter av påstander om at et produkt ikke inneholder genmodifisert materiale. Siden det er terskelverdier for sporforurensninger av EU-godkjent modifisert GMO, kan et produkt i visse tilfelle sies å være ikke-genmodifisert selv om det altså inneholder små forurensninger.
Alle genmodifiserte såvarer skal merkes med «Genmodifisert» i henhold til forskrift om såvarer28 § 29. Såvareområdet er innlemmet i EØS-avtalen, og merkebestemmelsen samsvarer med EUs bestemmelse.
6.4.5.4 Andre bestemmelser om genmodifisering under matloven
Bestemmelser om GMO er også gitt i andre forskrifter under matloven. Etter forskrift om økologiske landbruksprodukter mv.29, er det ved økologisk produksjon forbud mot bruk av GMO og produkter avledet fra slike organismer. Unntatt er genmodifiserte veterinærmedisiner. Det er altså krav til produksjonsmåten og ikke krav til selve produktet, noe som innebærer at det kan aksepteres sporforurensninger av genmodifisert materiale i et økologisk produkt. Økologiregelverket er implementert EU regelverk.
I plantesortsforskriften30 stilles det krav om at det skal framgå av norsk offisiell sortsliste om en sort er genmodifisert. Godkjente sorter i det enkelte EØS-land kommer på EUs felles sortsliste der det også skal gå fram om sorten er genmodifisert. Plantesortsforskriften er implementert EU-regelverk og er nært knyttet opp mot såvareforskriften.
I forskrift om plantehelse31 stilles det krav om at produksjon og omsetning av genmodifiserte planter og formeringsmateriale kun er tillatt dersom de er godkjent etter genteknologiloven. Samme forskrift stiller også krav om at slike planter og formeringsmateriale må være merket «genmodifisert». Plantehelseregelverket er nasjonalt regelverk og er ikke omfattet av EØS-avtalen.
6.4.5.5 Kontrollforskriften
EUs kontrollforordning samler alt kontrollregelverket i hele matkjeden og bidrar til å sikre én rettslig ramme for kontroll i hele matkjeden. Den skal sikre at europeiske myndigheter er harmoniserte, at de har systemer, organisering og tiltak på plass for å sikre etterlevelse av regelverket på matområdet. Reglene bidrar til forenkling og mer effektivt samarbeid mellom myndighetene. Forordningen gjelder offentlig kontroll av regelverket for mat, fôr, dyrehelse og dyrevelferd, animalske biprodukter, økologiske produkter og plantevernmidler. Rettsakten er gjennomført som kontrollforskriften under matloven, og ligger til grunn for størstedelen av Mattilsynets kontrollvirksomhet på matområdet i dag.
Mens kontrollforordningen gjelder fullt ut i EU, er det noen kontrollområder som ikke er en del av EØS-avtalen og som dermed ikke er underlagt tilsvarende rammeverk for kontroll i EØS/EFTA-statene. Dette gjelder områdene plantehelse, beskyttede betegnelser for næringsmidler og eksport til land utenfor EU-/EØS-området. Når det gjelder GMO på mat- og fôrområdet som er omfattet av EØS-avtalen, vil kontrollforordningen gjelde også for dette området når GM-pakken tas inn i EØS-avtalen. Selv om områdene ikke er innenfor EØS-avtalen, er det likevel bestemt at myndighetene må følge de samme prinsippene for risikobasert kontroll som fastsatt i kontrollforordningen, se omtale av offentlig kontroll i kapittel 6.4.6.
Kontrollforordningen omfatter også til dels utsettingsdirektivet, jamfør kapittel 6.4.2.6. Det er mulig å søke om kun dyrking av GMO til mat og fôr eller kun oppdrett av GMO produksjonsdyr. En slik produksjon vil kun være for eksport til tredje land, da mat og fôr må godkjennes etter GM mat- og fôrforordningen.
6.4.5.6 Særskilte beskyttelsestiltak
Det er særskilte beskyttelsestiltak ved import av ris og risprodukter fra Kina32, da det ved flere anledninger har blitt påvist ulovlig, ikke-godkjent genmodifisert ris i partier innført til flere EU-land. Alle forsendelser med opprinnelse i eller sendt fra Kina som inneholder ris eller produkter som inneholder ris, og hvor Norge er første mottaksstat, omfattes av denne forskriften. Mange produktgrupper/tollkoder omfattes av kravene om utvidet dokumentkontroll og analyser. Forskriften gjennomfører EUs regelverk om det samme, og er hjemlet i kontrollforordningen som i Norge er gjennomført i kontrollforskriften.
6.4.5.7 Regelverk om økt åpenhet
EUs åpenhetsforordning (forordning (EU) nr. 2019/1381; se kapittel 6.4.2.7) har medført endring i syv forskrifter33 under matloven og en paragraf i genteknologiloven (se kapittel 6.2.8). Da forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr ennå ikke er innlemmet i EØS-avtalen, er ikke den delen av åpenhetsforordningen gjennomført. Norge står likevel fritt til å fastsette tilsvarende nasjonale regler. Endringene i denne forordningen i EU er uansett nyttige også for norske matmyndigheter, fordi de får tilgang til den samme informasjonen som EFSA får fra søker.
6.4.5.8 Annet aktuelt regelverk – regler for sameksistens
Hvis det i en fremtidig situasjon blir godkjent GMO til dyrking i Norge, må det sikres at de som dyrker konvensjonelle eller økologiske avlinger ikke lider økonomiske tap eller opplever andre ulemper pga. innblanding av GMO i avlingene som dyrkes i nærheten av arealer hvor det dyrkes GMO, såkalt sameksistens. Utsettingsdirektivet hjemler retten til å gjennomføre nasjonale tiltak som sikrer dette.
Landbruks- og matdepartementet vurderte i 2006 at regelverk om sameksistens ligger innenfor både matlovens formål og saklige virkeområde, jamfør matloven §§ 1 og 2, og avklarte med daværende Miljøverndepartementet at ansvaret for oppfølgingen av sameksistens i Norge skulle ligge hos landbruks- og matmyndighetene.
Mattilsynet utarbeidet i 2007 utkast til nasjonalt regelverk om hvordan en eventuell fremtidig dyrking av genmodifiserte vekster skal foregå for i så liten grad som mulig å påvirke konvensjonelt og økologisk jordbruk – såkalt sameksistensregler for å hindre at konvensjonelle og økologiske avlinger blir kontaminert av GMO. Forskriftsutkastet ble utarbeidet med basis i EU Kommisjonens retningslinjer fra 2003 (rekommandasjon 2003/556/EF).
Utkastet til forskrift om dyrking mv. av genmodifiserte vekster foreslås med ett unntak bare gjort gjeldende for virksomheter. Unntaket er kravene til avstandsisolering, som foreslås gjort gjeldende for enhver som dyrker genmodifiserte vekster i nærheten av en virksomhet. Dyrkingsregler er utarbeidet for dyrking mv. av genmodifisert potet og mais, som de mest aktuelle arter i en fremtidig situasjon.
Forskriftsutkastet inneholder både generelle og artsspesifikke krav til dyrkingen. Eksempler på generelle krav er blant annet kravet om opplæring og autorisasjon, melde- og informasjonsplikt, merkekrav hvis vilkårene ikke følges, opplysningsplikt og overtakelse av forpliktelser. Eksempler på artsspesifikke krav er blant annet avstandsisolering, dyrkingsintervall, rengjøring av maskiner og utstyr samt etterkontroll.
Forskriftsutkastet går lenger enn både EU Kommisjonens retningslinjer fra 2003 og for eksempel Danmarks bestemmelser i det å ta hensyn til naboer av den som dyrker genmodifiserte vekster. I henhold til kommisjonens retningslinjer er sameksistens mellom ulike dyrkingsformer kun et spørsmål om økonomi. Bestemmelsene i Mattilsynets forskriftsutkast er imidlertid utformet slik at naboer til den som dyrker genmodifiserte vekster, ikke skal påføres noen ulemper, verken økonomiske eller andre.
Foreslåtte virkemidler er satt ut fra et mål om at det ikke skal forekomme noen spredning av genmodifisert materiale (<0,1 %) til nabovirksomhetenes avlinger. Dette er gjort av flere grunner. Ved å legge virkemidlene på et strengt nivå vil man for eksempel gi full frihet til nabovirksomheten til å ha en hvilken som helst produksjon vedkommende måtte ønske på hele sin eiendom. Ved å legge opp til ingen eller minimal spredning av genmodifisert plantemateriale til avlinger fra arealer der det ikke dyrkes genmodifiserte vekster, vil en også i større grad kunne se bort fra faren for økende andel av genmodifisert plantemateriale utover i produksjonskjeden som følge av en innblanding fra mange ulike kilder. En vil også ved å legge vilkårene på et strengt nivå redusere faren for opphoping av genmodifiserte frø i frøbanken i jorda, noe som kan gi problemer med opprettholdelse av sameksistens på lang sikt.
Mattilsynet har ikke formelt vurdert forskriftsutkastet av 2007 opp mot Kommisjonens nyere retningslinjer om sameksistens fra 2010. En uformell gjennomgang kan tyde på at de generelle kravene i forskriftsutkastet står seg opp mot de «nye» retningslinjene:
Forskriften ble utformet slik at konvensjonell og økologisk produksjon skulle sikres mot innblanding av GMO med en terskelverdi satt til «null» (dvs. deteksjonsgrensen).
Forskriftsutkastet ble grundig gjennomgått med referansegruppen.
Mattilsynet anbefalte at det ble laget en egen forskrift om kompensasjon rettet mot eventuelle skadelidte, og Statens Landbruksforvaltning laget i den forbindelse et forslag til kompensasjonsordning og finansiering av denne.
Etter Mattilsynets vurdering i 2007, er det ikke hjemmel i matloven til blant annet å innføre dyrkingsregler overfor andre enn næringsdrivende, pålegge private opplysningsplikt om dyrking eller kreve gebyr fra de som dyrker genmodifiserte vekster for å finansiere en kompensasjonsordning til naboer av disse som måtte lide et økonomisk tap som følge av uønsket innblanding av GMO i avlingen. Vurdert under ett, foreslo derfor Mattilsynet at det lages en ny bestemmelse i matloven om sameksistens som helt klart gir Kongen adgang til å fastsette forskrifter om alle forhold som angår dette – både gebyrinnkreving fra GM-dyrkerne, de ulike dyrkingsreglene og at alle bestemmelsene i forskriftene om ønskelig skal kunne gjøres gjeldende for både virksomheter og enhver.
6.4.6 Offentlig kontroll
6.4.6.1 Innledning
Virksomheter som importerer produkter med risiko for innblanding av genmodifisert materiale, skal ha rutiner i internkontrollsystemet sitt for å unngå dette. Mattilsynet fører tilsyn med at regelverket etterleves gjennom å kontrollere importerte mat-, fôr- og såvarer.
I Mattilsynets portefølje av overvåknings- og kartleggingsprogrammer, inngår programmet «Genmodifisering i mat, fôr og såvarer». Programmet har som formål å overvåke markedet og bidra til bevisstgjøring av industri og bransje med hensyn til regelverket og for behovet for dokumentasjon og internkontroll på området. Resultatene av den årlige kontrollen gir myndighetene informasjon om tilstanden på det norske markedet.
Utviklingen av dette tilsynsområdet startet opp for 23 år siden, og Mattilsynet har i dag god oversikt over produktgruppene med størst risiko for innblanding av genmodifisert materiale. De senere årene har Mattilsynet innhentet mer kunnskap om tilstanden også når det gjelder andre produktgrupper.
Mattilsynet fører tilsyn både etter matloven og genteknologiloven. Daværende Miljøverndepartementet delegerte tilsynsmyndighet etter genteknologiloven til Mattilsynet fra 2004. Begrunnelsen var sammenfallende interesser i matloven og genteknologiloven – en og samme handling kan innebære brudd på begge regelverk, at Mattilsynet har et stort tilsynsapparat som gjør tilsynet rasjonelt, og at det er sammenfallende tilsynsobjekter. Delegasjonen omfatter §§ 18-21 og § 24 og omfatter gjelder kontroll med om antatt formeringsdyktige produkter i gruppen av mat, fiskefôr og innsatsvarer i landbruket (fôr, såvare, plantedeler m.m.) inneholder GMO. Delegasjonen omfatter ikke forsøksfelt og levende GMO til annen bruk enn mat og fôr, herunder prydplanter, akvariefisk og andre hobby- og kjæledyr som Mattilsynet ellers fører tilsyn med etter annet regelverk i matloven. Ved funn av genmodifiserte organismer som er i strid med genteknologiloven skal Mattilsynet innhente miljøfaglig vurdering fra Miljødirektoratet før det vedtas reaksjon etter loven.
Mattilsynet skiller ikke på levende og prosessert mat og fôr i tilsynet med om virksomheten følger regelverket, men sammenfatter resultatene fra antatt formeringsdyktige produkter i et eget kapittel i den årlige rapporten.
Kontroll med genmodifisering er svært ressurs- og kompetansekrevende og byr på en rekke utfordringer, både for virksomheter, tilsynsmyndigheter og analyselaboratorier. I Mattilsynet er det satset på å bygge opp denne kompetansen ved utvalgte avdelingskontorer i regionene. Veterinærinstituttet er oppnevnt av Mattilsynet som nasjonalt referanselaboratorium på området, og er en viktig kunnskapsstøtte.
6.4.6.2 Risikobasert tilsyn og kontroll
Mattilsynet fokuserer både på analyser med tanke på eventuelt innhold av genmodifisert materiale, og vurdering av den dokumentasjonen virksomhetene besitter for å vise at regelverket etterleves. Virksomhetene må selv dokumentere at eventuelt innhold av genmodifisert materiale er under terskelverdiene, at innholdet er utilsiktet eller teknisk uunngåelig, og at det er godkjent i EU.
Det gjøres et risikobasert prøveuttak, hvor det i tillegg til planteart vektlegges eksportlandets status i forhold til dyrking av GMO. Fokus er lagt på importører, grossister og produsenter som håndterer produkter der genmodifisering er en relevant problemstilling. Råvarer av lite bearbeidete produkter prioriteres fordi slike produkter har best analyserbarhet. Årlig tas ut cirka 150 prøver for analyse og dokumentasjonskontroll, primært av produkter med innhold av soya, mais og raps.
I tillegg gjennomføres obligatorisk offentlig kontroll av ris importert fra Kina, oppfølging av bekymringsmeldinger og funn i forbindelse med annet tilsyn, samt eventuelle norske notifiseringer i EUs varslingssystem RASFF om funn av ikke EU-godkjente genmodifiserte produkter på markedet.
Generelt bekrefter de årlige resultatene at forekomsten av ulovlige genmodifiserte produkter i Norge er lav. Imidlertid er det vanskelig å unngå innblanding av sporforurensninger, på grunn av den globale markedssituasjonen. Dette gjelder mat og fôr, mens status for innførsel av såvare er meget god og uten påvisning av sporforurensninger.
Det er virksomhetens ansvar å risikovurdere alle varer som ønskes importert og å etablere nødvendige rutiner slik at norsk regelverk overholdes, også med hensyn til forekomst av ikke-godkjent genmodifisert materiale. Dette bør være en grunnleggende del av importørens internkontrollsystem. Den vanligste dokumentasjonen er sporbare analysesertifikater for sluttprodukter eller råvarer der genmodifisering er en risiko. Analysesertifikater skal være sporbare til aktuelt vareparti og av en viss kvalitet – blant annet må analysene være egnet til å påvise aktuelle forurensninger og utført av et laboratorium som er akkreditert for slike analyser.
I mange importerte mat- og fôrvarer, for eksempel i oljer, sukkerprodukter, tilsetningsstoffer som lecitin eller fôrmidler som maisgluten, er råvarene så bearbeidet at DNA i stor grad er ødelagt eller fjernet. Her vil analyser av sluttprodukt eller prosessert ingrediens/fôrmiddel som regel ikke kunne gi et godt svar med hensyn til innhold av genmodifisert materiale. For slike produkter må importøren innhente annen dokumentasjon, for eksempel sporbare analyser av råvarene som ingrediensene er produsert fra, eller IP-dokumentasjon (dokumentasjon på at råvaren er holdt adskilt i egen linje gjennom hele verdikjeden og med renholdsprotokoller, inspeksjonsrapporter og analysesertifikater i flere ledd fra såvare til ferdig produkt).
På matområdet har rundt 40 prosent av de kontrollerte importørene de senere årene hatt tilstrekkelig kunnskap og gode nok internkontrollrutiner for å unngå import av ulovlige genmodifiserte produkter. De største utfordringene har vært hos små og mellomstore virksomheter, blant annet frittstående dagligvareforretninger, spesialforretninger og innen netthandel. Det kommer stadig nye aktører på markedet. Det er likevel generelt få funn av produkter med ulovlig innhold av genmodifisert materiale.
Status for import av fôrråvarer er god, selv om det er vanskelig å unngå sporforurensninger av genmodifisert materiale. Kartlegginger i særlig 2017 og 2020 av import av fôrblandinger til hest, andre produksjonsdyr og kjæledyr, har vist et forbedringspotensial når det gjelder kunnskap og rutiner for å forebygge ulovlig innførsel av genmodifiserte produkter.
Såvaremarkedet er stort sett stabilt med hensyn til aktører og produkter, og internkontrollen og dokumentkvaliteten har blitt bedre de senere år.
Mattilsynet har stort fokus på veiledning av virksomheter slik at det skal bli enklere å gjøre rett. Det er viktig med tydelig og brukervennlig veiledningsmateriell som er lett tilgjengelig for alle aktuelle aktører.
6.5 Om deteksjonsmetoder og sporbarhet
6.5.1 Analytisk deteksjon og sporbarhet
Dagens regelverk om genmodifisert mat og fôr i EU, sikrer at tilsynsmyndighetene har tilgjengelige spesifikke analysemetoder for å kunne identifisere enhver EU-godkjent GMO. I Norge er disse analysemetodene viktige for tilsynsmyndigheten for å overvåke og kontrollere markedet for eventuelle ulovlige, genmodifiserte produkter, se nærmere beskrivelse av Mattilsynets offentlige kontroll med genmodifisering i kapittel 6.4.6.
De spesifikke analysemetodene er også nyttige for å ivareta krav om sporbarhet og merking, selv om generell sporing og merking også kan ivaretas uten slike analysemetoder. Ved hjelp av dokumentbasert sporbarhet skal man i prinsippet kunne følge produkter hele veien gjennom en produksjonskjede (jamfør kapittel 6.4.2.3 og 6.4.5.1.4), også dersom produktet etter hvert er sammensatt av elementer som har helt forskjellige opprinnelser. Analytisk sporbarhet er imidlertid viktig for å kunne verifisere renhet og avdekke udeklarerte og utilsiktede urenheter.
De generelle sporbarhetskravene som følger av regelverk med hjemmel i matloven, innebærer at enhver aktør i matkjeden fram til omsetning til forbruker er pålagt å kjenne opprinnelsen til en vare (dvs. ett ledd bakover) og å kunne dokumentere hvem som er kjøper eller mottaker av varen (dvs. ett ledd framover). Kravet gjelder for hele varen og også dersom varen er bearbeidet/ompakket og består av deler fra flere ulike leverandører/partier og/eller distribueres til flere mottakere.
Såkalt identity preservation (IP) har til formål å sikre at identiteten til et produkt kan spores gjennom hele verdikjeden, for eksempel fra såvare til butikk. IP-sikring brukes av mange produsenter for å sikre at det ikke innblandes ikke-godkjent GMO, og er spesielt aktuelt for eksport fra tredjeland til EU. Men IP-sikring er like aktuelt for virksomheter som for eksempel har avlet fram en spesielt verdifull plantesort eller dyrerase, og frykter at det underveis i verdikjeden kan skje en innblanding av mindre verdifulle produkter som utgis for å være den verdifulle typen. Det er for eksempel flere ganger dokumentert at verdifull hvitfisk som torsk er blitt erstattet av mindre verdifull hvitfisk i prosesserte matvarer på det europeiske marked. IP-sikring kan bidra til at en produsent har kontroll på at torsk ikke erstattes av annen mindre verdifull hvitfisk underveis i kjeden. Et GMO-relevant eksempel kunne vært genom-redigerte gråskimmelresistente jordbær dyrket uten sprøytemidler, som kanskje ville vært dyrere å produsere enn konvensjonelle jordbær, og hvor ønsket om å sikre seg mot innblanding av billige sprøytede jordbær ville forutsette bruk av IP og analyser som identifiserer den aktuelle resistente jordbærsorten.
Ønsket om å unngå GMO kan også føre til at man bygger opp IP rundt et system av analysemetoder som primært skal avdekke eventuell innblanding av GMO i en verdikjede. Det er en pragmatisk tilnærming dersom man ikke har et utgangspunkt som er genetisk unikt og homogent, men er samtidig svært krevende å drifte fordi det etter hvert finnes et svært stort og voksende utvalg av GMO som man må kunne fange opp analytisk hvis systemet skal være sikkert. IP egner seg generelt bedre for å dokumentere tilstedeværelse (evt. mengde) av noe unikt enn å dokumentere fravær av et bredt spekter av uønskete ting.
Analytisk sporbarhet forutsetter at det finnes en eller flere sporbare analytter, altså markører/egenskaper som kan påvises med analysemetode(r) og som representerer noe som på en unik måte karakteriserer produktet som skal spores, se figur 6.3 for flere detaljer. Det kan for eksempel være et genmodifiseringsspesifikt motiv i et gen i en GMO, eller et protein som gir en organisme en spesifikk egenskap. En sporbar analytt vil være et svært nyttig utgangspunkt for IP-sikring. Uten en sporbar analytt må sporbarhet baseres kun på dokumenter.
Figur 6.3 Sporbare analytter er en forutsetning for å kunne etablere analysemetoder. I prinsippet kan både DNA (inkludert metyleringsmønsteret), RNA, aminosyrer/proteiner og en del fenotypiske egenskaper danne utgangspunkt for analytisk sporbarhet (identifisering av en genetisk endret egenskap). Det er imidlertid stor forskjell på hvor godt egnet alternativene er. Mønsteret av basepar i DNA er det sikreste utgangspunktet og derfor gullstandarden for de metodene som alle de europeiske referanselaboratoriene benytter (European Network of GMO Laboratories; ENGL). Oversettelse fra DNA til RNA og videre til proteinsyntese og observerbare fenotypiske egenskaper, så vel som metyleringsmønster, påvirkes av en lang rekke miljøfaktorer. Både opprinnelse og mengde vil derfor være mindre sikre og stabile kilder til påvisning, identifisering og kvantifisering av spesifikke genteknologiavledete produkter, dersom man ikke benytter DNA-koden som utgangspunkt.
Figur: Arne Holst-Jensen
Generelt vil antall kopier av et gitt gen eller en annen DNA-sekvens være det samme i alle organismens celler, selv om det finnes noen unntak som ikke drøftes nærmere her. Avlesning til RNA og videre oversettelse til aminosyrer, proteinfolding og fenotypisk egenskap er generelt vesentlig mindre forutsigbar, noe som gjør at det i praksis kun er DNA-sekvens som egner seg som utgangspunkt for kvantifisering av genteknologiske produkter. RNA er et ustabilt molekyl og egner seg dårlig også for kvalitative analyser, mens proteinbaserte metoder er mye benyttet til hurtigtesting i store verdikjeder (for eksempel ved mottak og omlasting av større transporter med landbruksråvarer). Fenotypiske egenskaper som sprøytemiddelresistens er nyttige for eksempel for å teste visse såvarer. Det samme proteinet kan imidlertid stamme fra ulike DNA-sekvenser, noe som gjør at man ofte ikke kan si sikkert om det er en (spesifikk) genmodifisering som er kilden til et protein.
Analysekostnader varierer også betydelig, ikke minst vil det være av stor betydning hvor mye prøvemateriale som må analyseres for å oppnå et tilstrekkelig pålitelig analyseresultat, og hvor mange ulike typer av metoder og utstyr som kreves for å utføre analysearbeidet.
Boks 6.2 Om krav til analysemetoder
Forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr angir krav i forbindelse med søkers plikt til å levere en egnet analysemetode (se artiklene 3 og 17). Artikkel 32 og forordningens tillegg (Annex) redegjør for rollene til EURL-GMFF og ENGL. EURL-GMFF og ENGL har sammen utarbeidet en rekke veiledningsdokumenter som skal gjøre det enklere for søkere og saksbehandlere å sikre at søknader er fullstendige og at analysemetoder er egnet.1 Disse oppdateres og nye utarbeides ved behov.
Et av de sentrale veiledningsdokumentene redegjør spesifikt for hvilke krav selve analysemetoden må oppfylle2. Artikkel 2.1.2 Praktiserbarhet (practicability) understreker viktigheten av at metoden skal være praktiserbar på linje med andre anvendelser for tilsvarende formål. Praktiserbarhet defineres som graden av enkelhet av operasjonalisering, gjennomførbarhet og effektivitet ved bruk, og den tilknyttede unitære kostnaden (for eksempel kostnad per prøve) ved bruk. Det presiseres at en metode anses uakseptabel dersom den, uten tilstrekkelig saklig grunn, medfører (blant annet): nye typer av apparater som ikke er allment tilgjengelige eller særlig dyrt utstyr, ressursene som kreves for å utføre analysene (tid, arbeidsmengde, reagenser, kostnader) er vesentlig høyere enn det som kreves for analyser for tilsvarende formål, eller totalt analysevolum overstiger 25 µl. Andre praktiserbarhetsbetraktninger kan også medføre at en metode anses ikke-praktiserbar. Artikkel 2.3.1 omtaler krav til metodens spesifisitet, definert som metodens evne til å respondere eksklusivt til den aktuelle analytten. Her er akseptanskriteriet at spesifisiteten er dokumentert både in silico (mot relevante databaser o.l.) og eksperimentelt mot nærmere angitte mengder av materiale fra andre eventer, ikke-GMO-er og den aktuelle GMO-en.
Spesifikke deteksjons-, kvantifiserings- og toleransegrenser for GMO-innblanding inngår i Norges og EUs GMO-regelverk, så vel som i regelverk i mange andre land og kommersielle handelsavtaler. Flertallet viser til at teoretisk kan bortimot alle typer av genteknologiavledete produkter påvises (Bertheau 2019), men at i praksis vil få alternativer oppfylle praktiserbarhetsvilkårene som er beskrevet av EURL-GMFF og ENGL. For eksempel vil man kanskje være avhengig av å bruke spesielle analyseapparater og særlig ressurskrevende databehandlinger, i tillegg til at man kanskje må bruke opp urimelig mye av et produkt og betale en urimelig høy pris for å få svar som oppfyller regelverks- og/eller kontraktskrav.
1 https://gmo-crl.jrc.ec.europa.eu/guidance-documents
2 European Network of GMO Laboratories (ENGL). Definition of minimum performance requirements for analytical methods of GMO testing. 20 October 2015 (JRC95544). MPR Report Application 20_10_2015.pdf (europa.eu) (europa.eu). En ny veileder, som supplement til den gamle, er publisert i april 2023. Den supplerende veilederen finnes her: https://gmo-crl.jrc.ec.europa.eu/doc/JRC125975_01.pdf
EU har etablert et nettverk for GMO laboratorier i sine medlemsstater, samt Sveits, Norge og Tyrkia («European Network of GMO Laboratories» – ENGL). Hovedoppgaven er å støtte EUs referanselaboratorium for genmodifisert mat og fôr (EURL-GMFF) i dens oppgaver etter GM mat- og fôrforordningen, og å bidra til å løse utfordringene med påvisning, identifisering og kvantifisering av GMO.
Norge har deltatt i ENGL siden før den formelle opprettelsen i 2000, og Mattilsynet har oppnevnt Veterinærinstituttet som sitt nasjonale referanselaboratorium for analyser av genmodifisering i mat, fôr og såvarer. Både EURL-GMFF og ENGL har spesifikke roller i tilknytning til validering og implementering av offisielle analysemetoder for GMO i EØS-området, nærmere beskrevet i GM mat- og fôrforordningen og kontrollforordningen. Offisielle analysemetoder publiseres fortløpende på EURL-GMFFs nettsider34 hvor det også angis hvor relevant referansemateriale er tilgjengelig for referanselaboratoriene. Metodelisten omfatter også validerte analysemetoder som kan benyttes for screening m.m.
Boks 6.3 nedenfor, bestående av figurene 6.4–6.11 med forklarende tekst, viser ulike typer av genetiske endringer og mulighetene for å påvise disse analytisk.
Boks 6.3 Ulike typer av genetiske endringer og mulighetene for å påvise disse analytisk
Nedenfor beskrives seks sentrale typer av genetiske endringer, som spenner fra transgene GMO-er av den typen som har vært på verdensmarkedet i noen tiår, til de minste målrettede mutasjoner som dagens teknologi muliggjør. For å sette det i nødvendig kontekst er også endringer som oppnås uten bruk av genteknologi med. Med hvert eksempel kommenteres det også på hvilke DNA-baserte analytter (sekvensmotiver) man kan ha nytte av for å gjøre analytisk påvisning og identifisering. Avslutningsvis redegjøres det for hvordan man kan bruke kunnskap om egnede/tilgjengelige analytter til mer eller mindre kostnadseffektiv GMO-påvisning, og hvilke begrensninger både næringsaktører og kontrollmyndigheter har med tanke på analytisk sporbarhet.

Figur 6.4
Alle eksemplene starter med samme utgangspunkt, en organisme A (figur 6.4), som enten ønskes endret eller mistenkes å ha blitt endret. Alle individer av A, uansett om de er modifisert eller ikke, kan analytisk identifiseres med en endogen (artsspesifikk) sekvensmarkør (svart firkant). Slike sekvensmotiver brukes som referanse i GMO-analyser. Dels kvalitativt for å påvise at arten er tilstede i en prøve, dels for å kunne kvantifisere hvor mye av arten som er tilstede i prøven. Transgene GMO-er har en dominerende plass i GMO-sammenheng og er derfor et naturlig første eksempel.
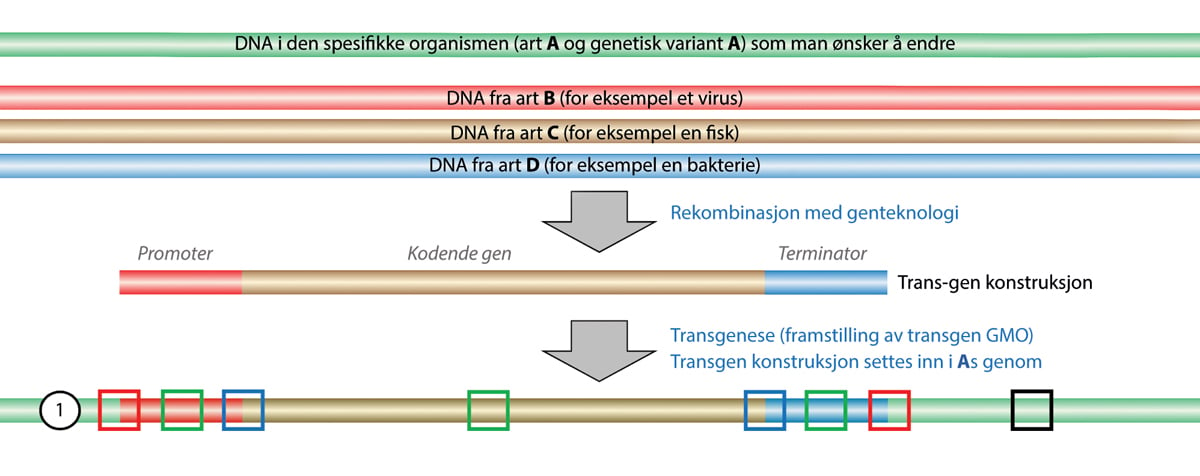
Figur 6.5
1. Bruk av rekombinant DNA-teknologi gjør at man kan hente ulike genetiske elementer og sette disse sammen til en ny genkonstruksjon (figur 6.5). Hvis elementene hentes fra en eller flere arter som ikke er «kryssbare» med organismen som skal endres, vil den nye konstruksjonen være transgen. I det øyeblikk konstruksjonen settes inn i organismen har man utført transgenese, og den modifiserte organismen er en transgen GMO. I en transgen GMO vil man ha mange ulike sekvensmotiver man kan benytte analytisk. Transgene elementer (grønne firkanter i figur 6.5) er elementer som ikke naturlig forekommer i den aktuelle arten. Fordi slike elementer som oftest har sin opprinnelse i en annen naturlig organisme og/eller kan være benyttet i mer enn en GMO, vil de som hovedregel være egnet som screening-markører eller potensielt bevis for at man har en GMO, men de vil bare unntaksvis kunne brukes for å identifisere en spesifikk GMO. Den transgene konstruksjonen er imidlertid satt sammen av flere transgene elementer, og elementer som ikke naturlig finnes i kombinasjon gir opphav til konstruksjonsspesifikke fusjonsmotiver (blå firkanter i figur 6.5). Disse vil kun finnes i GMO, og kan derfor brukes som bevis for at GMO er tilstede. Imidlertid er det ikke uvanlig at samme konstruksjon er benyttet i flere GMO-er. Konstruksjonsspesifikke motiver kan derfor ikke uten videre brukes til å bevise hvilken GMO som er tilstede. Når konstruksjonen settes inn i en ny organisme vil det med klassiske genmodifiseringsteknikker som setter inn nytt DNA på tilfeldig sted, oppstå unike fusjonsmotiver som er event- (hendelses-)spesifikke (røde firkanter i figur 6.5). Gullstandarden for GMO-påvisning er basert på slike motiver (Holst-Jensen et al. 2012) og det er koblingen mellom et slikt motiv og en unik identifikasjonskode (se boks 6.1) som er utgangspunktet for EUs analytiske sporbarhetssystemer (jamfør forordning (EF) nr. 1830/2003) om sporbarhets- og merkekrav.
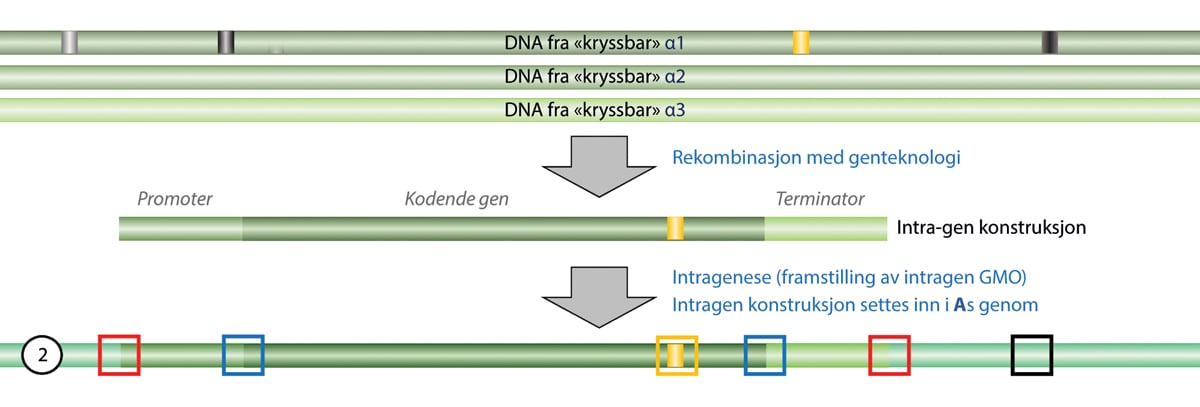
Figur 6.6
2. Rekombinant DNA-teknologi er også utgangspunktet for intragene konstruksjoner (se figur 6.6). Slike konstruksjoner består utelukkende av elementer som er hentet innenfor «kryssbare» organismer, men vil bestå av kombinasjoner av sekvensmotiver som ikke naturlig forekommer sammen i den gitte konstellasjonen. Det gjør at man i likhet med transgener får dannet konstruksjonsspesifikke sekvensmotiver (blå firkanter i figur 6.6). Og når en intragen konstruksjon settes inn på et tilfeldig/nytt sted i et genom vil det dannes eventspesifikke fusjonsmotiver (røde firkanter i figur 6.6). I eksemplet i denne figuren har man også ønsket å hente inn en mutasjon i et kodende gen (gul firkant) fra en av de «kryssbare» organismene. Denne mutasjonen finnes altså i den større «kryssbare» populasjonen, og påvisning av denne forteller derfor ikke med sikkerhet at man har å gjøre med en GMO, men den er en mulig screeningmarkør.
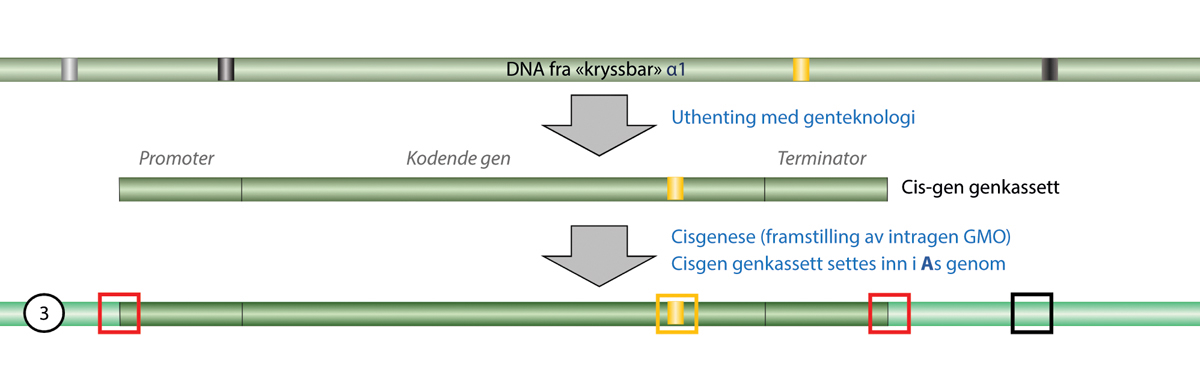
Figur 6.7
3. Cisgene genkassetter er komplette genkassetter som naturlig finnes i den «kryssbare» populasjonen, men kopieres og flyttes, og da enten kommer i tillegg til og/eller erstatter den tilsvarende genkassetten i organismen som endres (se figur 6.7). Man vil derfor få dannet eventspesifikke motiver (røde firkanter i figur 6.7) som kan benyttes til sikker påvisning og identifisering av GMO-en. I eksemplet i denne figuren er det en mutasjon (gul firkant) som er det attraktive grunnlaget for å lage en slik cisgen GMO. Denne mutasjonen finnes også i den større «kryssbare» populasjonen, og påvisning av denne forteller derfor ikke med sikkerhet at man har å gjøre med en GMO, men den er en mulig screeningmarkør.
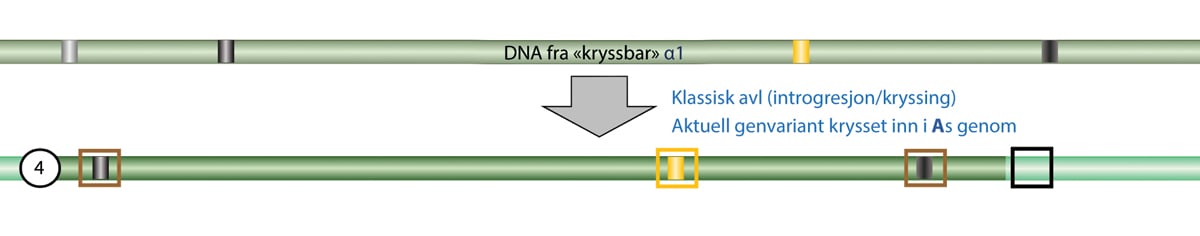
Figur 6.8
4. Innenfor «kryssbare» populasjoner vil det finnes genetisk variasjon som kan være attraktiv å flytte inn i en framforedlet genetisk bakgrunn (i figur 6.8 markert som mutasjonen i gul firkant). Med klassisk avl gjøres dette ved kryssing (introgresjon). Det vil da typisk følge med mer genetisk variasjon enn den man spesifikt ønsker å krysse inn (her vist som mutasjoner i brune firkanter). Ingen av disse motivene skyldes da genmodifisering.
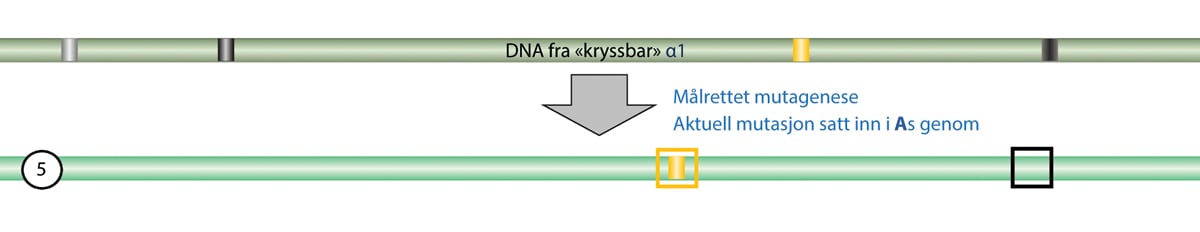
Figur 6.9
5. Med moderne genteknologiske verktøy kan man gjøre målrettet mutagenese, som tilsvarer å innføre en helt spesifikk mutasjon på et helt spesifikt sted i et genom (se figur 6.9). I dette eksemplet er en slik mutasjon (gul firkant) hentet fra en annen «kryssbar» organisme og satt inn i organismen man ønsker å endre. Siden denne mutasjonen også finnes naturlig i «kryssbar» populasjon vil det ikke være mulig å fastslå med sikkerhet at tilstedeværelse skyldes genmodifisering. Dersom mutasjonen ikke finnes naturlig, men er oppstått ved bruk av genteknologi vil man være avhengig av at utvikler eller noen andre offentliggjør at motivet har en slik opprinnelse, for at man med rimelig grad av sikkerhet skal kunne fastslå at motivet skyldes genmodifisering. I mange land i verden er slike mutasjoner ikke regulert som GMO, og produktet vil da kunne markedsføres uten at kjøpere vil ha tilgang til informasjon om mutasjonens opprinnelse.
Figur 6.10
6. Delesjoner (at et stykke av DNA tas bort) kan både skje naturlig og ved bruk av genteknologi (se figur 6.10). I begge tilfeller vil fragmenter spleises sammen slik at det oppstår et helt nytt fusjonsmotiv (rosa skråstilte firkanter i figuren; de og grønne ender fusjoneres). Hvis motivet er et resultat av genmodifisering, vil motivet være spesifikt for den aktuelle genmodifiseringen (eventspesifikt). For å kunne vite sikkert at motivet er oppstått ved bruk av genmodifisering er det imidlertid en forutsetning at noen (som regel utvikler selv) offentliggjør at motivet har en slik opprinnelse. Det vil i så fall ha samme status som andre eventspesifikke motiver (rød firkant i de øvrige figurene/eksemplene). Alternativt vil det ha samme status som en vilkårlig mutasjon (som grå og gule firkanter i de øvrige eksemplene).
Figur 6.11
Når man skal undersøke om et produkt er eller inneholder GMO, har man altså et utvalg av ulike sekvensmotiver som kan brukes som utgangspunkt for analysene (se figur 6.11). Endogen (artsspesifikk) markør (svart firkant) benyttes for å fastslå tilstedeværelsen og eventuelt kvantifisere mengden av den aktuelle arten. Screening for tilstedeværelse av motiver som finnes i mange GMO-er (grønne og gule firkanter) kan også være et godt verktøy for å gjøre analyser kostnadseffektive. Ulempen med disse er at de også forekommer naturlig, og derfor ikke med sikkerhet fastslår at GMO er tilstede. Fravær av slike motiver i en analyse er heller ikke sikkert bevis for at GMO ikke er tilstede. Slik screening benyttes imidlertid i utstrakt grad både av næringsaktører og kontrollmyndigheter for å velge ut/prioritere om en konkret prøve skal analyseres grundigere. Også konstruksjonsspesifikke motiver (blå firkanter) inngår i screeningstrategier, og fordelen med disse er at de med sikkerhet fastslår at GMO er tilstede. Gullstandarden er likevel de eventspesifikke motivene (rød firkant, og evt. rosa firkant) som helt spesifikt indentifiserer hvilken GMO som er tilstede og gjør det mulig å kvantifisere mengden relativt til den artsspesifikke markøren (svart firkant). Med målrettede mutagenese- og genomredigeringsteknikker vil man i mange tilfeller stå overfor scenarier av type 5 (se figur 6.9) og 6 (se figur 6.10), og disse vil ikke med sikkerhet kunne skilles fra scenarier av type 4 (se figur 6.8) eller på annen måte identifiseres sikkert som et resultat av genmodifisering.
Figurer: Arne Holst-Jensen
Mindretallet ønsker å presisere følgende til Boks 6.3
Teksten går ut over formålet med kapittel 6 «Dagens regulering og forvaltning av genmodifiserte og avledede produkter». Mye av teksten er vurderinger som bygger på svært forenklede eksempler, i kontrast til eksemplene i kapittel 7.
Boks 6.3, punkt 2 (intragenese). Eksempelet som er benyttet er noe villedende. Normalt vil en ønsket mutasjon frambringes på andre måter, f.eks ved cisgenese eller målrettet mutagenese. Det mest relevante aspektet i en intragen tilnærming er mulighet for nye kombinasjoner av genetiske elementer, herunder de som regulerer genet(ene). En intragen tilnærming vil derfor normalt gi flere rekombinasjonsseter (fusjonsmotiver, sekvensoverganger mellom de ulike genfragmentene) og kan heller ikke reproduseres ved konvensjonell foredling. Å henvise til informasjonsnivået i en mutasjon blir i dette tilfellet misvisende med hensyn på deteksjonsmuligheter, særlig siden eksempelet også viser at promotor og terminator er hentet fra andre kryssbare arter. Eksempelet viser heller ikke at disse regulatoriske elementene vil/kan være fra andre gener og gener som reguleres på andre måter. Eksempelet viser heller ikke til at «gullstandarden» (punkt 1) normalt vil være tilgjengelig.
Boks 6.3, punkt 3 (cisgenese). Mange av de samme kommentarene som for punkt 2 gjelder også her. For planter vil det i mange tilfeller dreie seg om allelvarianter og genetisk variasjon som ikke forekommer i matkjeden (EFSA, 2012), for eksempel ved å hente inn egenskaper fra viltvoksende slektninger som ikke er en del av matkjeden (introgresjon). Eksempelet viser ikke til forventet grad av genetisk variasjon i cisgener til bruk i dyreavl versus planteforedling. Eksempelet viser heller ikke til at «gullstandarden» (punkt 1) normalt vil være tilgjengelig.
Boks 6.3. punkt 4 (klassisk avl). Eksempelet dreier seg ikke om «Dagens regulering og forvaltning av genmodifiserte og avledede produkter» og fremstår noe villedende i denne sammenheng når det også kombineres med andre forenklede eksempler.
Boks 6.3. punkt 5 (målrettet mutagenese). Punkt 5 presenterer målrettet mutagenese, dvs. bruk av genteknologiske verktøy for spesifikt å endre én eller flere enkeltbaser i et genom. Det er i dag ikke en begrensning på antall endringer som innføres eller antall steder som endres i genomet. Omfanget kan være alt fra en enkeltnukleotidendring til flere endringer i rekke, til mange endringer ulike steder i et genom. Bakgrunnen for å innføre slike nukleotidendringer kan (som vist i eksempelet) være det å oppnå en kjent biologisk effekt av slik variasjon fra samme eller andre arter, eller etter ny utprøving/utvikling. I dette tilfellet baserer man seg på informasjon om sekvensvariasjon, og ikke fysisk DNA-materiale. Disse motivene (enkeltendringer eller flere genetiske endringer) kan derfor i noen tilfeller finnes naturlig i samme art eller en «kryssbar» populasjon, i andre tilfeller ikke. I tilfeller der nukleotidvariasjonen finnes naturlig i produkter som allerede er i matkjeden, vil det være utfordrende å identifisere en genmodifisering. På en annen side, dersom nukleotidendringene ikke finnes naturlig i andre produkter i matkjeden og er en sammenstilling av mange enkeltnukleotidendringer, vil det forventes at de kan påvises analytisk. Deteksjon av ulike genmodifiseringer er et aktivt forskningsfelt og det er ikke avklart om og hvordan dette vil tilnærmes fremover. Analytisk sporbarhet er heller ikke en forutsetning for dagens dokumentbaserte sporbarhet i matkjeden. Punkt 5 viser til at påvisning vil være vanskelig hvis ikke type genmodifisering er kjent. Dette gjelder imidlertid alle typer genetiske modifiseringer (punktene 1–5). Det er en forutsetning for rutinemessig analyse at type og omfang av genmodifisering er kjent. Dette gjelder også GMO i dag, da protokollene som benyttes er validert og basert på kjent type og omfang av genmodifisering. Med hensyn til analytisk sporbarhet av dagens kommersielle GMO, er det noen typer regulatoriske DNA-elementer som benyttes ofte som utgangspunkt for påvisningen, noe som kan effektivisere den analytiske tilnærmingen.
Boks 6.3 punkt 6 (delesjoner). Ny genomredigeringsteknologi kan også spesifikt fjerne DNA-områder i et genom. Dette medfører at gjenværende DNA-tråder spleises sammen slik at det oppstår et helt nytt fusjonsmotiv (sekvensen som representerer overgangen mellom de ulike DNA-fragmentene). Motivet vil være spesifikt for den aktuelle genmodifiseringen (eventspesifikt) siden samme delesjonsmotiv ikke vil kunne reproduseres ved konvensjonelle metoder. Eksempelet viser heller ikke til at «gullstandarden» (punkt 1) normalt vil være tilgjengelig også her.
Mindretallet mener at en konklusjon av gjennomgangen av ulike typer genmodifisering ved bruk av nye genteknologier, illustrerer at 4 av 5 tilnærminger i hovedsak lar seg detektere analytisk. For målrettet mutagenese vil mulighet for deteksjon måtte avklares videre.
Flertallet viser til at i mars 2019, publiserte ENGL rapporten «Detection of food and feed plant products obtained by new mutagenesis techniques»35. De konkluderer her med at det er høyst usannsynlig at laboratorier skal kunne oppdage tilstedeværelse av uautoriserte planteprodukter med små DNA-endringer som enkeltbaseendringer eller korte indels, ved hjelp av dagens etablerte og rutinemessig brukte deteksjonsmetoder og analyseinstrumenter. Slike mat- eller fôrprodukter, kan derfor komme inn i EU uoppdaget, dersom man ikke mottar forutgående informasjon om de endrede DNA-sekvensene.
Mindretallet viser til at for noen genredigeringsteknikker (målrettet mutagenese) er det foreløpig ikke utviklet gode verktøy for detektering. Dette viser at teknologien på dette området foreløpig er umoden. Det er derfor viktig å utvikle pålitelige metoder som blir praktisk tilgjengelige og som er økonomisk akseptable for aktørene i matkjeden.
Mindretallet viser til at i mars 2019 publiserte det europeiske nettverket av GMO-laboratorier ENGL en rapport om utfordringer knyttet til deteksjon av planter til mat eller fôr utviklet ved nye genomteknikker. ENGL konkluderte med at det var usannsynlig at en genomredigert plante med enkeltbaseendringer eller korte indels vil kunne detekteres på en tilfredstillende måte etter gjeldende regelverk. Problemet er særlig knyttet til å kunne påvise med sikkerhet at en endring i DNA er resultat av en genmodifisering. ENGL skrev også at det ikke var etablert prosedyrer som legger til rette for en entydig konklusjon om at genomredigering har skapt endringen. ENGL konkluderte også med at disse utfordringene ikke gjaldt for genomredigerte planter med mer omfattende endringer i arvestoffet.
Mindretallet viser videre til at i EUs niende rammeprogram for forskning og innovasjon, Horisont Europa, ble det i 2022 igangsatt et forskningsprogram for nye deteksjonsmetoder for produkter utviklet ved bruk av nye genomteknikker36. Formålet er å gi myndigheter, utviklere og aktører i matkjeden deteksjonsverktøy for å sikre åpenhet og pålitelighet. Dette vil i neste omgang bidra til å sikre forbrukere retten til å gjøre informerte valg. Programmet skal bidra til utvikling og validering av deteksjonsmetoder for produkter oppnådd gjennom nye genomiske teknikker. Budsjettrammen er om lag 100 millioner kroner (10 millioner euro), hvorav 50 millioner kroner finansieres av Europakommisjonen. Det foregår også forskning på deteksjonsmetoder i Norge, blant annet gjennom prosjektet FoodPrint, finansiert av Norges forskningsråd.37
6.5.2 Analytisk deteksjon og sporbarhet for genredigerte produkter
Fravær av sporbar analytt kan være et problem gjennom hele eller sene deler av verdikjeden. Dersom analytten er et innsatt DNA-motiv eller et spesifikt protein, så kan bearbeiding av en råvare for eksempel føre til at DNA og/eller protein blir fjernet eller nedbrutt. Dette har man lang erfaring med, for eksempel med planteoljer og sterkt varmebehandlete produkter fra transgene GMO-er som har vært på verdensmarkedet i flere tiår.
Dagens EU-regelverk om genmodifisert mat og fôr krever merking og sporbarhet av produkter som er fremstilt fra GMO, uavhengig av om det er påviselig analytt i produktet på det aktuelle punktet i verdikjeden. Dokumentbasert sporbarhet er derfor nødvendig for implementering av dagens regelverk. Man har så langt ingen erfaring med produkter hvor en sporbar analytt mangler gjennom hele verdikjeden. Med målrettede mutagenese- og genomredigeringsteknikker kan det utvikles produkter hvor genteknologi er benyttet under framstillingen, men intet nytt genetisk motiv er til stede i den levende organismen man sitter med til slutt (boks 6.3).
Som tidligere nevnt, sikrer krav i EUs regelverk om genmodifisert mat og fôr tilgang til eventspesifikke analysemetoder for påvisning og kvantifisering av alle GMO som søkes godkjent i EU som mat og fôr. Kravet om spesifikke analysemetoder innebærer at en søker ikke vil få godkjent den aktuelle GMO-en dersom det ikke leveres en slik spesifikk analysemetode sammen med et egnet referansemateriale (prøvemateriale av den aktuelle GMO-en som kan benyttes som kontroll ved bruk av metoden), samt dokumentasjon som skal vise at metoden oppfyller et sett med kvalitetskrav som beskrevet i retningslinjer fra EURL-GMFF og ENGL.38 For de minst omfattende målrettede mutagenesebaserte endringene vil det, i henhold til ENGL-rapporten etter dagens kunnskap ikke være mulig å lage en analysemetode som kan oppfylle disse kravene. Det kan etter dagens regelverk medføre at slike produkter ikke kan få godkjenning.
6.5.3 Sporbarhet ved import av genredigerte produkter
De fleste GMO har ett eller flere handelsnavn, som benyttes i markedsføring og andre sammenhenger. Dette kan bidra til å skape forvirring om hva det siktes til. For å gjøre det enkelt å sikre at man snakker om den samme GMO-en, gis hver enkelt GMO en unik identifiseringskode (se kapittel 6.4.2.3). Det er imidlertid ikke alle land i verden som har krav om at dette systemet skal benyttes. Land som ikke definerer alle typer målrettede endringer innen art som genmodifisering vil da heller ikke ha krav om at slike organismer skal få en unik identifiseringskode.
6.5.4 Påvisningsmuligheter for produkter som ikke omfattes av GM mat- og fôrregelverket
Flertallet viser til at utsettingsdirektivet som gjelder levende GMO som ikke er til mat og fôr, krever at søker opplyser om «metoder til påvisning, identifikation og – hvis det er relevant – kvantificering af transformationsbegivenheden», samt metoder til sporing av GMO-ene. Flertallet mener at forholdet mellom matregelverket og utsettingsdirektivet kompliseres av at enhver anvendelse må være godkjent, samtidig som det alltid vil være en viss sannsynlighet for at godkjenning ikke er gitt for et aktuelt formål. Eksempler er avskårne lilla nelliker, som i dag er de eneste godkjente GMO-ene i Norge. Nellikene er ikke godkjent for humant konsum, men Interflora oppfordrer til å benytte kronblader fra blant annet nellik som pynt på maten.39 Andre eksempler er prosesserte produkter som verken skal dyrkes eller spises av dyr eller mennesker, for eksempel bomullstekstiler eller papir fra genmodifiserte trær, som i dag ikke krever noen form for godkjenning og heller ikke omfattes av krav til merking, sporbarhet eller analytisk påviselighet. Flertallet viser til at enkelte kan hevde at det strider imot de verdibaserte intensjonene bak dagens genteknologilov med hensyn til bærekraft, samfunnsnytte og etikk, og ikke gir forbrukere eller andre noen mulighet til å gjøre verdibaserte valg. Også slike produkter kan dessuten ende opp som fôr eller mat, for eksempel i en mer sirkulær bioøkonomi hvor bomull og papir kan være fôrråvare for avfallsbasert insektproduksjon.
6.5.5 Event, trait, unique identifier og stack
Et stort antall av GMO-ene som i dag er godkjent som mat og fôr i EU, er såkalte stacked-events (se boks 6.1). Det vil si GMO hvor flere modifiserte gener og egenskaper er kombinert. For disse har EU så langt hatt en inkonsistent praktisering av kravet til spesifikk analysemetode og identifisering gjennom mat- og fôrkjeden. Boks 6.1 inkluderer et eksempel på stacked events: krysningen av de fire GMO-maisene SYN-BTØ11-1 × SYN-IR162-4 × SYN-IR6Ø4-5 × MON-ØØØ21-9.
Allerede noen av de første genmodifiserte produktene på markedet hadde flere tilførte egenskaper. Dels var det vanlig at man hadde tilført en seleksjonsmarkør, slik som et gen som koder for resistens mot spesifikke antibiotika. Dels var det tilført gen for minst én, men også tidvis flere egenskaper som man ønsket at organismen skulle kunne uttrykke i kommersiell bruk. Eksempelvis inneholder mais Bt176, en av de første kommersielle GMO-maisene (godkjent i EU i 1997), både en antibiotikaresistensmarkør (bla), et gen som gir toleranse for glufosinat-ammonium herbicider (bar), og et gen som koder for insektresistens (cry1Ab). Denne maisen ble genmodifisert med bruk av genkanon. I de fleste faglige sammenhenger omtales også slike GMO-er som stacks. Bt176 har imidlertid status som én event (SYN-EV176-9). Bt176 er derfor et eksempel på stacked traits (kombinasjon av egenskaper), men ikke på stacked events.
Produkter utviklet med de nyere genteknologiene vil på samme måte kunne inneholde kombinasjoner av endrede og/eller nye gener, framstilt enten samtidig eller ved krysning av produkter med ulike endrede gener. Det kan være hensiktsmessig å omtale også slike som stacks. Derimot er endringen med nyere genteknologi ikke alltid en transformasjon i klassisk forstand, så begrepet event må eventuelt omdefineres for noen slike produkter. Fordi slike produkter heller ikke defineres som GMO i flere jurisdiksjoner utenfor EU/EØS vil de heller ikke nødvendigvis bli tildelt en unik kode som kan benyttes i forbindelse med informasjonsutveksling, sporbarhet, m.m.
Det er flere grunner til å se nærmere på fenomenet stacks. Dette er dels knyttet til hvordan de risikovurderes, men også knyttet til spørsmål som godkjenningsstatus, analytisk identifiserbarhet og merkekrav (Taverniers et al. 2008).
Ved for eksempel en søknad til EU om godkjenning av maishybriden Bt11 x MIR162 x MIR604 x GA21, må EFSA vurdere de ulike kombinasjonene (det er totalt 4 kombinasjoner av tre av eventene; 6 kombinasjoner av to av disse og selvfølgelig også hver av de fire individuelle eventene).
I en populasjon som i utgangspunktet består av en spesifikk stack (gjelder både stacked events og stacked traits) vil det alltid være en viss sannsynlighet for at de ulike modifiserte genene segregerer i forbindelse med celledelinger og når det produseres avkom, kjønnet eller ukjønnet. EUs regelverk stiller også krav om at hver event skal kunne identifiseres med sin egen spesifikke og validerte analysemetode. I EU vil en event vil ikke bli godkjent uten slik metode, inkludert tilgang til relevant referansemateriale. For eksempelet med maishybriden Bt11 x MIR162 x MIR604 x GA21, anser man at man har en egnet analysemetode fordi man har en analysemetode for hver av de fire eventene som ble krysset. Selv om de eventuelt re-segregerer vil man kunne påvise spesifikke eventer. Analysemetoden er imidlertid spesifikk for ett konkret DNA-sekvensmotiv i den enkelte event, og dette sekvensmotivet kan i noen tilfeller også segregere bort.
Bt11 inneholder to traits/egenskaper (pat og cry1Ab), MIR162 inneholder én trait i tillegg til seleksjonsmarkør (vip3Aa20 og pmi) i likhet med MIR604 (mcry3A og pmi), mens GA21 innholder én trait (mepsps). For hver av disse eventene er det kun én trait som påvises med den offisielle analysemetoden. I maiseventen Bt176 er det satt inn to traits og en seleksjonsmarkør, men også i denne eventen er det kun én trait som påvises med den offisielle analysemetoden. Bildet blir mer komplisert, fordi en trait kan forekomme i mer enn én kopi i en event. Det er for eksempel tilfelle i Bt176. Det medfører at man kan ha avkom av Bt176 som har én eller begge traits, men uten at denne GMO-en kan påvises med den offisielle analysemetoden.
EUs regelverk har krav om at alle godkjente GMO mat- og fôrvarer som utilsiktet eller uunngåelig inneholder eller består av mer enn 0,9 % GMO eller prosessert GMO, per ingrediens, skal merkes. Lenge var det en diskusjon om dette skulle forstås med henvisning til genetisk mengde eller produktmasse (Holst-Jensen et al. 2006). Stacks kompliserte bildet, fordi man analytisk ikke vil kunne skille mellom samtidig tilstedeværelse av for eksempel de fire eventene Bt11, MIR162, MIR604 og GA21, og tilstedeværelse av krysningen av alle fire. Analysemetodene kvantifiserer mengden GMO som andel genetisk mengde. Dagens EU-regelverk angir at dette skal oversettes til produktmasse. I praksis innebærer dette et stort tolkningsrom, fordi man enten kan velge å tolke mengden av påvist Bt11 + MIR162 + MIR604 + GA21 som summen av fire ulike GMO-er som er til stede i produktet samtidig, eller som påvisning av en stacked event som består av alle fire. Eksempelvis, hvis alle fire påvises i en mengde tilsvarende 0,5 % av maisen i en matvare, vil den første tolkningen resultere i at GMO-innholdet måles til 2 %, mens den siste tolkningen vil resultere i at GMO-innholdet måles til (4 x 0,5 %)/4 = 0,5 %. Dagens terskelverdi for krav til merking i EU og aksept i visse tilfeller av sporforurensning av EU-godkjente GMO i produkter i Norge, er satt til 0,9 %. Dette vil kunne medføre vesentlige konsekvenser. Virksomheten som eier produktet vil kunne hevde at produktet inneholder en stacked event, mens en som er sterkt opptatt av merking vil kunne hevde at virksomheten må bevise dette for å slippe unna merkekrav. Hvis virksomheten kan bevise noe slikt før produktet analyseres, så blir det i neste omgang et spørsmål om tilstedeværelsen er utilsiktet eller teknisk uunngåelig, siden informasjonen jo da kan antas å ha vært tilgjengelig før produktet ble omsatt. Vilkårene for å slippe merking i EU, og vilkåret for å slippe å søke om godkjenning i Norge, er i dag i begge tilfeller at tilstedeværelsen er utilsiktet og teknisk uunngåelig. Disse problemstillingene er bare delvis avklart i dagens regelverk og tilsynspraksis.
Problemstillingene beskrevet over, vil også gjelde for produkter utviklet med nye genteknologier.
I Mattilsynets offentlige kontroll vurderes GMO-analyseresultater fra sak til sak. I saker der det kan være tvil om påvist innhold av genmodifisering i et produkt kommer fra en stack eller fra flere enkelt-eventer, blir Veterinærinstituttet koblet inn i vurderingen av analyseresultatet. Mattilsynets setter dessuten en tilstrekkelig høy margin på analyseresultatet for å ta høyde for utfordringen med kvantifiseringen, slik at det ikke skal være noen tvil i de tilfeller der det påvises ulovlig GMO i et produkt. I tillegg blir alltid annen dokumentasjon for produktet som er analysert, innhentet og vurdert før det eventuelt fattes et vedtak.
6.6 Tilgrensende matlovsregelverk – bruk av dyr i forsøk
6.6.1 Innledning
Før en virksomhet kan søke om godkjenning for klinisk utprøving av et legemiddel, gjøres en rekke forsøk fra grunnforskning til «pre-pre-kliniske» og pre-kliniske forsøk. Dataene innsamlet i forbindelse med grunnforskning og frem til og med pre-kliniske forsøk hvor det brukes forsøksdyr, er basis for senere søknad om klinisk utprøving etter legemiddellovgivningen og etter genteknologiloven dersom legemidlene består av GMO (se nærmere omtale i kapittel 6.3).
For å kunne bruke dyr i grunnforskning og senere pre-kliniske forsøk, må man få tillatelse av Mattilsynet før igangsettelse av forsøket. Enhver virksomhet som driver med forsøksdyr, må også forhåndsgodkjennes av Mattilsynet.
Klinisk utprøving av veterinære legemidler på dyr er stadiet etter forsøksstadiet. Klinisk utprøving på dyr i normale besetninger/husholdninger gjøres for å bekrefte data som stammer fra studier på forsøksdyr, for å dokumentere at tiltenkt doseringsregime, bruksmåte m.v. fungerer i praksis ved forskjellige typer dyrehold og andre ulike forhold. I dokumentasjonen til søknader om klinisk utprøving av legemidler til dyr finnes det derfor data fra laboratoriestudier og pre-kliniske forsøk på samme dyreart (målarten), hvor det allerede er gjort ulike vurderinger mht. dyrevelferd, dyrehelse, etikk og samfunnsnytte. Virksomheter som søker om klinisk utprøving, har med basis i pre-kliniske forsøk fått vitenskapelige data som tilsier at de veterinære legemidlene kan prøves ut på dyr i normale besetninger/husholdninger. Utprøvingen gjøres for å bekrefte data som stammer fra de pre-kliniske forsøkene, for å dokumentere at tiltenkt doseringsregime, bruksmåte m.v. fungerer i praksis ved forskjellige typer dyrehold og andre ulike forhold. Hvis den kliniske utprøvingen har vært på matproduserende dyr, kan dyrene etter utprøvingen er ferdig og etter en eventuell tilbakeholdelsestid for legemiddelet, som oftest gå inn i matkjeden. Dette til forskjell fra forsøksdyr brukt i grunnforskning og pre-kliniske forsøk, hvor dyrene oftest avlives og skrottene destrueres ved godkjente destruksjonsanlegg etter at forsøkene er ferdige.
6.6.2 Forsøksdyrforskriften
1. Formål og virkeområde
Bruken av dyr i forsøk reguleres av Forskrift 18. juni 2015 nr. 761 om bruk av dyr i forsøk (Forskrift om bruk av dyr i forsøk – Lovdata). Forskriften er hjemlet i dyrevelferdsloven, dyrehelsepersonelloven og matloven. Formålet med forskriften, jamfør § 1, er å begrense bruken av dyr til vitenskapelige og utdanningsmessige formål, fremme god velferd og respekt for dyr som brukes til slike formål, og bidra til at dyrene ikke utsettes for unødige belastninger.
§ 2 sier at forskriften gjelder når dyr
a) blir brukt eller er ment å bli brukt i forsøk eller
b) blir oppdrettet spesielt for at deres organer eller vev kan bli brukt til vitenskapelige formål.
Forskriften gjelder levende virveldyr, tifotkreps og blekksprut. Dette omfatter også tidlige utviklingsstadier av disse dyrene hvis sanseapparatet er på et tilsvarende nivå som hos ferdig utviklede dyr, herunder fostre av pattedyr i siste tredjedel av normal utvikling og larver av virveldyr som ernærer seg selv. I tillegg gjelder forskriften når dyr på enda tidligere utviklingsstadier blir brukt i forsøk og får leve videre og sannsynligvis vil oppleve smerte, frykt, varig skade eller annen belastning etter å ha nådd utviklingsstadier som nevnt i annet punktum.
Forskriften gjelder selv om det brukes beroligende, bedøvende eller smertestillende midler, eller andre metoder slik at dyret ikke påføres smerte, frykt, varig skade eller annen belastning.
Forskriften gjelder inntil dyr som nevnt i første ledd er avlivet, omplassert eller tilbakeført til et dyrehold.
Unntatt fra forskriften er likevel blant annet:
klinisk utprøving av legemidler til dyr når dette er nødvendig for å få eller beholde markedsføringstillatelse.
Klinisk utprøving er ikke å anse som forsøk. Søknad om klinisk utprøving av legemidler til dyr behandles av legemiddelmyndighetene og evt. også miljømyndighetene hvis utprøvingen gjelder utsetting av levende GMO. Unntatt fra forskriften er også:
handlinger som det ikke er grunn til å tro vil påføre dyret smerte, frykt, varig skade eller annen belastning tilsvarende eller større enn ved å føre inn en nål etter god veterinær praksis.
Kravene i forskriften er rettet mot oppdrettere, formidlere og brukere.
2. Definisjoner og unntak
Det er ingen særskilte krav i forskriften vedrørende grunnforskning og pre-kliniske forsøk med GMO-legemidler til human eller veterinær bruk, og slike forsøk faller derfor inn under ‘medisinsk virksomhet’ og reguleres på samme måte som annen bruk av dyr i forsøk.
Forsøk slik det er definert i § 4, omfatter også etablering og vedlikehold av genmodifiserte dyrestammer som kan påføre dyret belastninger.
3. Krav om godkjenning
Det følger av § 5 at oppdrettere, formidlere og brukere, og lokalene de bruker, skal være godkjent av Mattilsynet. Videre følger det av § 6 at Dyr kan brukes i forsøk bare hvis Mattilsynet har godkjent forsøket. Det er ikke lov å benytte dyr i forsøk hvis man kan oppnå samme kunnskap uten bruk av dyr. Det skal alltid benyttes så få dyr som mulig, og forsøket skal alltid utføres slik at dyrevelferden er best mulig under forsøket.
Forsøk med dyr klassifiseres i lett, moderat eller betydelig belastende for forsøksdyrene. Vedlegg B nevner eksempler i de ulike klassene. I klassen ‘lett belastende’ forsøk, finner vi blant annet
Avl av genmodifiserte dyr hvor effekten på fenotyper forventes å være mild
Farmakokinetiske studier hvor det gis en enkelt dose av en substans og tas et begrenset antall blodprøver (totalt < 10 % av sirkulerende blodvolum), og substansen ikke forventes å gi noen påvisbar skadelig virkning
Administrering av stoffer subkutant, intramuskulært, intraperitonealt, intravenøst via overfladiske blodkar og via sonde, hvor stoffet kun har en mild innvirkning på dyret, og volumene er innenfor passende grenser med hensyn til dyrets størrelse og art
I klasse ‘moderat belastende forsøk’, er aktuelle eksempler blant annet
Hyppig bruk av testsubstanser som gir moderate kliniske effekter, og uttak av blodprøver (> 10 % av sirkulerende blodvolum) på bevisste dyr i løpet av få dager uten at volumtapet erstattes.
Studier for fastsettelse av akuttoksiske doser, tester for kronisk toksisitet/carsinogenitet med ikke-dødelige endepunkter
Avl av genetisk modifiserte dyr, som forventes å resultere i en moderat skadelig fenotype
Etablering av genetisk modifiserte dyr gjennom kirurgiske inngrep
I gruppen ‘betydelig belastende forsøk’, er aktuelle eksempler blant annet
Toksisitetstesting med døden som endepunkt eller hvor dødsfall må forventes og det fremkalles alvorlige patofysiologiske tilstander (for eksempel akuttoksisitetstesting av en enkelt dose (se OECDs retningslinjer for testing))
Utprøving av vaksine karakterisert med vedvarende svekkelse av dyrets tilstand, progressiv sykdom som fører til døden, forbundet med langvarig moderat smerte, frykt eller annen belastning
4. Krav til forsøkene
Søknaden til Mattilsynet skal ha et forsøkssammendrag, jamfør § 8, som skal være lett forståelig for allmennheten og bla. beskrive forsøkets formål og forventet vitenskapelig eller samfunnsmessig nytteverdi, samt forventede skadevirkninger på dyrene og om hvordan kravene til erstatning, reduksjon og forbedring skal etterleves.
Reglene om å bruke alternative metoder enn dyreforsøk, å bruke så få dyr som mulig og å forbedre metodene for å minske belastningen på forsøksdyrene, jamfør blant annet §§ 9-11, er et hovedformål i forsøksdyrforskriften. Disse prinsippene kalles også 3R: Erstatning (Replacement), Reduksjon (Reduction) og Forbedring (Refinement). 3R er etisk betinget: Det følger av § 24 og vedlegg E, at de som arbeider med dyr skal ha tilstrekkelig utdanning og praksis, herunder om etikk relatert til forholdet mellom mennesker og dyr, livets egenverdi og argumenter for og imot bruk av dyr til vitenskapelige formål.
Forsøksdyr er levende vesener med evnen til å føle frykt og smerte. Det er i alles interesse å redusere lidelsen til et absolutt minimum, også fordi dyr som er frie for smerter og stress vil de gi de mest pålitelige resultatene i et forsøk. Det er derfor både gode etiske og vitenskapelige grunner til å behandle dyrene så godt som mulig.
Forskriften inneholder en rekke bestemmelser for å sikre forsøksdyrenes velferd og stiller kompetansekrav til personale som planlegger eller utfører dyreforsøk samt til personell som steller og håndterer dyr. Forskriften har videre en rekke vedlegg som gir detaljerte bestemmelser for innholdet i søknader, klassifisering av forsøk etter belastningsgrad, avlivningsmetoder, utdanningskrav, krav til hold av dyr m.m.
6.6.3 Mattilsynets vurdering av søknader om bruk av forsøksdyr
Forsøksdyrforskriften og Instruks om Mattilsynets forvaltning av forsøksdyrforskriften40, gjennomfører til sammen direktiv 2010/63/EU om bruk av dyr til vitenskapelige formål. Mattilsynet skal godkjenne alle virksomheter som driver med forsøksdyr i Norge, og behandler de enkelte søknadene om forsøk og bruken av forsøksdyr. For søknader om bruk av genmodifiserte forsøksdyr til utsetting i miljøet, forutsetter Mattilsynet at Miljødirektoratet har godkjent forsøksutsettingen. Ved arbeid med genmodifiserte dyr eller arbeid med genmodifiserte mikroorganismer i kombinasjon med dyr, må det sendes en melding eller søknad om godkjenning (avhengig av GMO-ens risikoklasse) om innesluttet bruk og kombinasjonsbruk til Helsedirektoratet. Kopi av utfylt meldeskjema må legges ved i den aktuelle FOTS-søknaden41 til Mattilsynet.
Hver søknad om dyreforsøk blir vurdert ut fra en kost-/nyttevurdering der de 3R-ene er helt sentrale, og alle søkerne må greie ut om disse i søknadsskjemaet. Mattilsynet har oppnevnt seks eksperter innenfor 3R til å hjelpe til med innspill i behandling av søknader. Regjeringen har dessuten opprettet en Nasjonal komité for beskyttelse av forsøksdyr (forsøksdyrkomitéen) i tråd med EUs forsøksdirektiv. Denne komiteen skal være et rådgivende organ for forvaltningen og de enkelte forsøksdyrvirksomhetene. Formålet er å fremme en harmonisert implementering og utveksling av informasjon og best practice om de tre R-er, dyrevelferd og etikk på området.
Planer om dyreforsøk må kvalitetssikres på minst tre ulike nivåer: de skal være lovlige, de skal være av høy vitenskapelig kvalitet og de skal være etisk forsvarlige. Det siste er naturlig nok det vanskeligste, men vitenskapelige standarder er heller ikke alltid lett å enes om. For å veie opp verdien av et forsøk mot belastningen på dyret, må det foretas en nytte-kostnadsanalyse. Forsøkets samfunnsnytte samt etiske forhold rundt bruken av forsøksdyr, er viktige prinsipper som vurderes av Mattilsynet i forbindelse med godkjenning av søknad om bruk av forsøksdyr.
Den nasjonale forskningsetiske komite for naturvitenskap og teknologi (NENT) har utarbeidet etiske retningslinjer for bruk av dyr i forskning42, med formål om å gi etisk veiledning til forskere og andre som vurderer dyreforsøk. Mye av innholdet i retningslinjene tilsvarer krav nedfelt i forsøksdyrforskriften, men de skisserer også etiske spørsmål forbundet med bruk av genmodifiserte dyr i forskning. Videre beskrives ansvaret forskeren har for å følge føre-var-prinsippet når det foreligger troverdig, men usikker kunnskap mht. om dyreforsøk kan føre til uakseptable konsekvenser for dyrebestander og økosystemer som helhet.
6.6.4 Instruks om Mattilsynets forvaltning av forsøksdyrforskriften
Når Mattilsynet mottar en søknad om bruk av dyr i forsøk, gjøres en grundig vurdering av om forsøket
a) er vitenskapelig, utdannelsesmessig eller medisinsk begrunnet, eller er påbudt
b) har et formål som berettiger bruk av dyr
c) er tilrettelagt slik at forsøkene kan utføres på den mest mulig skånsomme og miljøvennlige måten
d) vil oppfylle øvrige krav i forsøksdyrforskriften.
Videre skal Mattilsynet vurdere
a) forsøkets formål, særlig de forventede vitenskapelige gevinstene eller den utdannelsesmessige eller medisinske verdien
b) om forsøket er i samsvar med kravet om erstatning, reduksjon og forbedring i forsøksdyrforskriften § 9
c) klassifisering av forsøkene etter belastning i samsvar med forsøksdyrforskriften vedlegg B
d) analysere forsøkets skadevirkninger og gevinster for å vurdere om dyrenes lidelse, smerte og frykt kan berettiges av de forventede resultater med hensyn til etiske betraktninger, og om virkningene i siste omgang kan gavne mennesker, dyr eller miljøet
e) bedømme grunnlaget for unntak fra hovedreglene i forsøksdyrforskriften § 12 om lokalisering, § 14 om bedøvelse og smertebehandling, § 16 om avliving, § 17 om gjenbruk av dyr, § 19 om truede dyrearter, § 20 om primater, § 22 om dyr som skal være avlet for forsøk, § 23 om eierløse og forvillede dyr av domestiserte arter, og § 29 om levemiljø og stell
f) avgjøre om og når forsøket skal evalueres etter forsøksdyrinstruksen § 13.
Det følger av instruksen at Mattilsynet skal sørge for at søknaden om godkjenning av forsøk blir vurdert av personer med særskilt kompetanse innen
a) de vitenskapelige fagområdene som er relevante for forsøket, blant annet kompetanse om erstatning, reduksjon og forbedring av forsøk
b) utforming av forsøk, blant annet kompetanse om nødvendig statistikk
c) veterinær forsøksdyrpraksis, eventuelt veterinær viltpraksis
d) hold eller håndtering av de dyreartene som skal brukes.
6.6.5 Bruk av forsøksdyr i Norge43
I 2022 var det 90 godkjente virksomheter som driver med forsøksdyr i Norge44, hvor dyr holdes og brukes til forskning og undervisning. I tillegg gjøres det forsøk med ville dyr i felt. I 2022 ble det tilsammen rapportert 1 443 130 dyr brukt i godkjente dyreforsøk.45 Av disse var cirka 95 % fisk, hvorav laks utgjorde den klart største gruppen. Fisk dekker et stort forskningsområde innen akvakultur-, bestands-, biomedisinsk- og basalforskning, men antall fisk varierer mye fra år til år. Dette skyldes at antallet store forsøk i oppdrettsnæringen varierer.
Av pattedyr ble det i 2022 brukt flest mus (53 817 individer). Mus og rotter brukes først og fremst i medisinsk grunnforskning relatert til humane sykdommer, som for eksempel genmodifiserte mus som brukes i kreftforskning og annen medisinsk forskning. Men selv om det overordnete målet vil være å «finne en ny og bedre medisin mot en sykdom» er de fleste forsøkene på et så tidlig stadium i prosessen at det ikke kan kalles pre-klinisk utprøving, men defineres som grunnforskning. På fisk stiller det seg annerledes, da forskningen her gjøres på arten legemiddelet er ment til bruk for og veien frem til legemiddel er kortere.
Fire hunder ble i 2022 brukt i et terminalt forsøk, men andre forsøk på hund og andre arter var i kategorien minimal og opp til lett belastning.
Årsrapportene fra forsøksdyrforvaltningen i Mattilsynet viser et økende forbruk av mus og en nedgang i bruk av rotter som forsøksdyr det siste tiåret.46 Dette skyldes økende tilgang på genredigerte mus som nyttige modellorganismer for ulike menneskesykdommer. Genredigerte zebrafisk er også modellorganismer som i økende grad tas i bruk både for humanmedisin og veterinærmedisin. Den store og mer varierende bruken av fisk skyldes hovedsakelig forskning som kan gi økt kunnskap om oppdrettsfiskenes biologi og mestring av forholdene i oppdrettsanleggene og ny teknologi, samt testing av nye vaksiner og medisiner (pre-kliniske forsøk).
Søknader til Mattilsynet har de siste årene ligget på i underkant av 600 per år, som fordeler seg på rundt 450 søknader om forsøk og resten om endringer i allerede godkjente forsøk og søknader om godkjenning av virksomheter. Søknadene dreier seg om forskning på sykdommer, utvikling av medisiner og dyrefôr, samt ulike adferdsstudier.
Det er generelt få avslag på søknader, anslagsvis 5-10 per år. Det lave antallet avslag skyldes ikke at det er enkelt å få tillatelser, men at det er både dyrt og komplisert å gjøre dyreforsøk slik at de søknadene som kommer stort sett er velbegrunnede og sendt fra høyt kompetente fagfolk. I tillegg har Mattilsynet lagt seg på en forvaltningslinje der søker kontaktes og veiledes ved uklarheter eller mangler i søknaden. Svært få søknader blir godkjent uten denne dialogen og veiledningen.
I tillegg til Mattilsynets behandling av søknader, føres tilsyn med virksomhetene som utfører dyreforsøk, herunder selve dyreholdet, dyrestallen, dyrepasserne og øvrig personell.
6.6.6 Vurdering av etikk, samfunnsnytte og bærekraft for søknader om bruk av forsøksdyr
Dataene innsamlet i forbindelse med grunnforskning og pre-kliniske forsøk hvor det brukes forsøksdyr, er basis for senere søknad om kliniske utprøving etter legemiddellovgivningen. Det er altså småskalaforsøkene som må gjennomføres før full-skala klinisk utprøving, som vurderes av Mattilsynet i forbindelse med tillatelser etter forsøksdyrforskriften. Klinisk utprøving gjøres sent i utviklingsløpet og omfattes av legemiddellovgivningen, og er ikke søknadspliktig som dyreforsøk.
Forsøksdyrforskriften §§ 8-11, § 24 og vedlegg E hjemler vurdering av samfunnsmessig nytteverdi og etikk for ethvert forsøk som innebærer bruk av dyr i grunnforskning og pre-kliniske forsøk. ‘Bærekraftig utvikling’ er ikke omtalt i forskriften eller Mattilsynets instruks, men bærekraftvurderinger er gjort i enkeltstående tilfeller. Det kan anføres at veterinære legemidler generelt har som formål å redusere sykdom. Foruten at redusert sykdom fremmer dyrevelferd, så fremmes bærekraft ved at tap av dyr under produksjonsprosessen reduseres. Dette fører til behov for færre dyr og redusert belastning på klima og natur. Ifølge FAO er friske dyr mer produktive og genererer lavere utslipp per produktenhet.
Mattilsynets behandling av søknader om bruk av forsøksdyr gjøres av personell med særskilt fagkompetanse, som beskrevet i egen instruks. Grunnforskningen og de pre-kliniske forsøkene Mattilsynet har gitt tillatelse til, er vurdert av Mattilsynet til å være etisk forsvarlige og ha en positiv samfunnsnytte. De store full-skala forsøkene som klinisk utprøving representerer, gjennomføres på de samme arter og ofte under de samme forhold som små-skala forsøkene hvor Mattilsynet har vurdert samfunnsnytte og etikk (for eksempel utsetting av fisk i merder; mindre forsøksmerder kontra store, kommersielle).
6.7 Føre-var-prinsippet
6.7.1 Innledning
I det følgende omtales føre-var-prinsippet særskilt. Prinsippet gjelder i utgangspunktet for de fleste områder, men har fått spesiell oppmerksomhet når det gjelder søknader om godkjenning av GMO.
Føre-var-prinsippet gir føringer for hvordan man i forbindelse med beslutninger og ulike handlinger skal håndtere manglende kunnskap og vitenskapelig usikkerhet. Prinsippet har som utgangspunkt at det er bedre å forebygge at vesentlige skader inntreffer, enn å prøve å «reparere» i ettertid. Prinsippet blir ofte omtalt som et prinsipp som skal la tvilen komme miljøet og naturen til gode. Det innebærer at man skal unngå vesentlig skade på naturen og miljøet når man fatter beslutninger, og at manglende kunnskap ikke skal brukes som begrunnelse for å unnlate å treffe tiltak.
Prinsippet kan ha ulik benevnelse uten at det materielle innhold er ment å være forskjellig. I Danmark og Sverige omtales det som et forsiktighetsprinsipp, mens det i engelsktalende land gjerne omtales som «precautionary principle» eller «precautionary approach».
Det virkelige gjennomslaget for føre-var-prinsippet var under FNs Rio-konferanse i 1992. På dette toppmøtet ble både klimakonvensjonen og konvensjonen om biologisk mangfold undertegnet. Senere er føre-var-prinsippet utviklet videre gjennom et samspill mellom norsk og internasjonal rett. Prinsippet inngår nå i en rekke traktater, bade globale og regionale, som Norge er tilsluttet. Prinsippet følger også implisitt av Grunnloven § 112. Videre er prinsippet lovfestet i norsk rett gjennom blant annet naturmangfoldloven av 2009 § 9, Svalbardmiljøloven av 2001 § 7 og havressursloven av 2008 § 7.
Rio-erklæringen prinsipp 15:
«In order to protect the environment, the precautionary approach shall be widely applied by states according to their capabilities. Where there are threats of serious or irreversible damage, lack of full scientific certainty shall not be used as a reason for postponing cost effective measures to prevent environmental degradation.»
Føre-var-prinsippet etter naturmangfoldloven § 9 gjelder som en retningslinje ved utøving av offentlig myndighet, jamfør naturmangfoldloven § 7. Prinsippet vil altså gjelde som en retningslinje også for saker etter genteknologiloven, når sakene har betydning for naturmangfoldet.
Naturmangfoldloven § 9 lyder:
«Når det treffes en beslutning uten at det foreligger tilstrekkelig kunnskap om hvilke virkninger den kan ha for naturmiljøet, skal det tas sikte på å unngå mulig vesentlig skade på naturmangfoldet. Foreligger en risiko for alvorlig eller irreversibel skade på naturmangfoldet, skal ikke mangel på kunnskap brukes som begrunnelse for å utsette eller unnlate å treffe forvaltningstiltak.»
Prinsippet legges i praksis også til grunn på mange områder der det ikke er lovfestet, og det er et sentralt hensyn bak ulike regelverk. I norsk lovgivning anvendes føre-var-prinsippet både i forvaltningen av genteknologiloven og matloven. I motsetning til regelverk i EU og etter Cartagena-protokollen, nedfelles imidlertid ikke prinsippet direkte i noen av de to lovene, men fremkommer som et resultat av en tolkning der rettskildefaktorer som forarbeidene, formål bak lovene og forvaltningspraksis er tillagt særlig vekt.
6.7.2 Føre-var-prinsippet i internasjonalt GMO-regelverk og EØS-regelverk
Cartagena-protokollen om biosikkerhet
Cartagena-protokollen er utformet i tråd med føre-var-prinsippet, som gjenspeilt i artikkel 1 om formålet til protokollen:
«In accordance with the precautionary approach contained in Principle 15 of the Rio Declaration on Environment and Development, the objective of this Protocol is to contribute to ensuring an adequate level of protection in the field of the safe transfer, handling and use of living modified organisms resulting from modern biotechnology that may have adverse effects on the conservation and sustainable use of biological diversity, taking also into account risks to human health, and specifically focusing on transboundary movements.»
Prinsippet er traktatfestet i EU med Maastrichttraktaten og nedfelt i traktaten om Den europeiske unions virkemåte (Lisboatraktaten) artikkel 191 (2). Prinsippet er også nedfelt i flere rettsakter, se ytterligere omtale i kapittel 5.5.1.
Utsettingsdirektivet 2001/18/EF
EUs direktiv 2001/18 om utsetting av GMO til miljøet er tatt inn i norsk rett gjennom genteknologiloven. Direktivet henviser til føre-var-prinsippet i fortalen punkt 8:
«Det er tatt hensyn til føre-var-prinsippet ved utarbeidingen av dette direktiv, og det må også tas hensyn til når det gjennomføres.»
Prinsippet er også direkte tatt inn i artikkel 4(1) om alminnelige bestemmelser:
«1. Medlemsstatene skal i samsvar med føre-var-prinsippet påse at alle egnede tiltak treffes for å unngå at utsetting eller markedsføring av GMO-er medfører skadevirkninger for menneskers helse og miljøet. GMO-er skal bare utsettes i miljøet eller markedsføres i samsvar med henholdsvis del B eller del C.»
Utsettingsdirektivets krav til en forhåndsvurdering av helse- og miljøeffekter, og overvåkning av GMO-en etter at den er satt ut i miljøet, er eksempler på at regelverket er utformet i tråd med føre-var-prinsippet.
EU-domstolen la i 2018 også vekt på direktivets utforming omkring føre-var-prinsippet i sin vurdering av om nye mutageneseteknikker burde unntas direktivets virkeområde på lik linje med konvensjonelle mutageneseteknikker (Sak C-528/16 om nye mutageneseteknikker).
General Food Law
Food Law er gjennomført i norsk rett gjennom matlovsforskriften med hjemmel i matloven. Føre-var-prinsippet som regulert i Food Law er derfor gjeldende rett i Norge. I fortalen punkt 21 heter det at:
«… i de særlige tilfellene der det er fare for liv eller helse, men der det råder vitenskapelig usikkerhet, er føre-var-prinsippet et middel til å fastsette risikohåndteringstiltak eller andre tiltak for å sikre det høye helsevernnivået som Fellesskapet har valgt.»
Artiklene 6 og 7 regulerer føre-var-prinsippet slik:
«Artikkel 6 Risikoanalyse
1. For å nå det allmenne mål om et høyt vernenivå for menneskers liv og helse, skal næringsmiddelregelverket bygge på risikoanalyser, unntatt når dette ikke er hensiktsmessig på grunn av omstendighetene eller tiltakets art.
2. Risikovurderingen skal bygge på tilgjengelige vitenskapelige bevis og utføres på en uavhengig og objektiv måte som gir innsyn.
3. Ved risikohåndteringen skal det tas hensyn til resultatene fra risikovurderingen, og særlig til uttalelsene fra myndigheten omhandlet i artikkel 22, andre faktorer av betydning for det aktuelle tilfellet og føre-var-prinsippet, når vilkårene fastsatt i artikkel 7 nr. 1 er relevante, med henblikk på å oppnå næringsmiddelregelverkets allmenne mål, som er fastsatt i artikkel 5.
Artikkel 7 Føre-var-prinsippet
1. I særlige tilfeller der det på bakgrunn av en vurdering av tilgjengelige opplysninger påvises en mulighet for skadelige virkninger på helsen, men der det fortsatt råder vitenskapelig usikkerhet, kan det vedtas midlertidige risikohåndteringstiltak som er nødvendige for å sikre det høye helsevernnivået som Fellesskapet har valgt, i påvente av ytterligere vitenskapelige opplysninger med sikte på en mer omfattende risikovurdering.
2. Tiltak som vedtas i henhold til nr. 1, skal stå i forhold til målet og ikke hindre handelen mer enn nødvendig for å sikre det høye helsevernnivået som Fellesskapet har valgt, samtidig som det tas hensyn til teknisk og økonomisk gjennomførbarhet og andre faktorer som anses å være berettigede for den aktuelle saken. Tiltakene skal vurderes på nytt innen et rimelig tidsrom avhengig av arten av den risiko for liv og helse som er påvist, og typen vitenskapelige opplysninger som er nødvendige for å avklare den vitenskapelige usikkerheten og foreta en mer omfattende risikovurdering.»
6.7.3 Om føre-var-prinsippet i genteknologiloven
Genteknologiloven har en oppbygning i tråd med føre-var-prinsippet. Det skal gjøres en forhåndsvurdering av konsekvenser for helse og miljø ved framstilling og bruk av GMO.
Prinsippet er ikke nevnt i selve lovteksten til genteknologiloven, men er omtalt flere steder i Ot.prp. nr. 8 (1992–93) Om lov om framstilling og bruk av genmodifiserte organismer.
I proposisjonen side 46 heter det:
«I høringsnotatet ble føre-var-prinsippet tatt opp under omtalen av bærekraftig utvikling av bioteknologi. Det ble uttalt at «prinsippet innebærer i denne sammenheng at der det er tvil om bruk av bioteknologi kan ha negative konsekvenser for miljø, helse eller andre forhold, bør tvilen komme natur og samfunn til gode». Flere høringsinstanser har gitt sin støtte til at prinsippet skal være et viktig utgangspunkt for lovgivningen. Prinsippet vil være særlig viktig ved utsettingssaker. UNIGEN mener at prinsippet er formulert for strengt, og dette syn har også etter høringen kommet fram i noen debatter der lovutkastet ble omtalt. Departementet vil understreke at føre-var-prinsippet ikke innebærer at all bruk av genteknologi i utgangspunktet anses som risikabelt. Men der det etter en konkret vurdering antas å være en rimelig grad av tvil om risiko, taler dette mot bruken. Føre-var-prinsippet vil gjenspeile seg konkret gjennom for eksempel krav om konsekvensutredning ved utsetting i miljøet, og ved skjønnsutøvelsen etter loven».
Prinsippet omtales igjen på side 49, i forbindelse med etikkvurderingen som skal gjøres etter loven. Her heter det:
«Vurdering av etiske konsekvenser må tillegges stor vekt ved all framstilling og bruk av genmodifiserte organismer, men det har sine begrensninger i det det i prinsippet forutsetter at en er i stand til å fremskaffe og vurdere alle tenkelige konsekvenser. (…) Det er derfor sentralt at en tar i bruk føre-var-prinsippet som blant annet medfører at der det er rimelig grad av tvil om tiltakets risiko, skal tvilen komme natur og samfunn til gode.»
Videre nevnes det på side 50, i forbindelse med omtalen av bærekraft. Her heter det:
«Et overordnet prinsipp i Brundtlandkommisjonens rapport er ønsket om å forebygge fremfor å reparere, det såkalte føre-var-prinsippet. Dette prinsippet innebærer i denne sammenheng at der det er rimelig grad av tvil om bruk av bioteknologi kan ha negative konsekvenser for miljø eller helse, bør tvilen komme natur og samfunn til gode.»
Prinsippet omtales også i forbindelse med de særskilte merknadene til lovens formålsparagraf, § 1. Her heter det blant annet:
«Når lovens formålsbestemmelse bruker ordet «uten» helse- og miljømessige skadevirkninger, må dette ikke forstås helt bokstavelig i det enkelte, konkrete tilfelle. (…) Begrepet «uten» skadevirkninger er likevel brukt for å understreke siktemålet om å vurdere risiko for helse og miljø på forhånd og å unngå mulige skadevirkninger, og om at føre-var-prinsippet skal legges til grunn.»
Videre omtales prinsippet i de særskilte merknadene til § 10 om godkjenning. Her heter det:
«Særlig når det gjelder utsetting i miljøet, må det legges vekt på føre-var-prinsippet. Rimelig grad av tvil om hvilken risiko for skade som kan oppstå, skal i utgangspunktet lede til at godkjenning ikke gis.»
Føre-var-prinsippet og forskrifter til genteknologiloven
Det følger av genteknologiloven § 11 at søknad om godkjenning av en utsetting etter genteknologiloven § 10 skal inneholde konsekvensutredning for å klarlegge risikoen for helse- og miljømessige skadevirkninger og andre følger av utsettingen.
Som følge av dette er det fastsatt en forskrift 16. desember 2005 nr. 1495 om konsekvensutredning etter genteknologiloven. Føre-var-prinsippet er omtalt i vedlegg 2 og vedlegg 4 til forskriften.
Vedlegg 2 inneholder prinsipper for miljørisikovurdering i henhold til forskriften § 13 – § 16. I del B redegjøres det for ulike generelle prinsipper som skal følges når miljørisikovurderingen foretas. Det vises videre til at dette skal skje «i samsvar med føre-var-prinsippet».
Vedlegg 4 omhandler vurdering av etikk, bærekraft og samfunnsnytte i henhold til forskriften § 17. Her er føre-var-prinsippet et av punktene som skal inngå i vurderingen. I del A listes følgende kontrollspørsmål som gjelder prinsippet:
«Er det rimelig grad av tvil om de foreliggende risikovurderingene, og er det fare for større risiko?
Er det rimelig grad av tvil om de foreliggende sannsynlighetsvurderinger, og er det fare for høyere sannsynligheter for skade?
Er det rimelig grad av tvil om de foreliggende konsekvensvurderinger, og er det fare for mer alvorlige konsekvenser, for helse eller miljø?
Er det rimelig grad av tvil om mulige, alvorlige kumulative konsekvenser for helse eller miljø?
Er det rimelig grad av tvil om foreslåtte modererende tiltak og virkemidler virker som forutsatt?»
Det fremgår av del B at hvis svaret er ja på ett eller flere av disse spørsmålene, så tilsier dette at søknaden kan avslås med henvisning til føre-var-prinsippet.»
Føre-var-prinsippet og forvaltningspraksis etter genteknologiloven
Søknad om godkjenning av en utsetting etter genteknologiloven § 10 skal inneholde konsekvensutredning for å klarlegge risikoen for helse- og miljømessige skadevirkninger og andre følger av utsettingen. Myndighetene vurderer om føre-var-prinsippet skal komme til anvendelse.
Som det heter i Ot.prp. nr. 8 (1992–93) innebærer prinsippet at der det er rimelig grad av tvil om tiltakets risiko, skal tvilen komme natur og samfunn til gode. «Rimelig grad av tvil» innebærer at noe usikkerhet aksepteres. Dette vil blant annet henge sammen med hvor alvorlige følger det er snakk om. Mindre usikkerhet aksepteres der det er snakk om potensielt alvorlige følger.
Føre-var-prinsippet er sentralt i vurderingene som gjøres av om det skal gis tillatelse til utsetting, men ikke avgjørende. Det foretas en konkret samlet vurdering, innenfor rammen av genteknologiloven § 10. Her vil som nevnt graden av usikkerhet og hvor alvorlige de potensielle negative følgene er, være viktig. Videre vil man se på kriteriene etikk, samfunnsnytte og bærekraft. I flere saker der miljømyndighetene har vært restriktive ut fra en føre-var-tankegang, har for eksempel samfunnsnytten vært begrenset.
Et eksempel som delvis kan illustrere dette er forbudet ved Kgl.res. 2. juni 2017 mot de genmodifiserte rapslinjene Ms8, Rf3 og Ms8xRf3. Regjeringen la avgjørende vekt på at rapslinjene kan utgjøre en miljørisiko. Både ikke-neglisjerbar risiko og føre-var-prinsippet ble lagt til grunn for forbudet. Samtidig ble rapslinjene ansett å ha lav samfunnsnytte. Det var ingen etterspørsel eller behov for rapslinjene i Norge. Rapslinjene hadde ikke egenskaper som gjorde dem bedre egnet enn annen raps for norske forbrukere. I kgl.res. legges det også vekt på at markedsføring av rapslinjene Ms8, Rf3 og Ms8xRf3 vil kreve at det iverksettes tiltak for å hindre frøspill og øvrige tiltak i hele verdikjeden. Saken er omtalt nærmere i kapittel 8 og kapittel 10.
6.7.4 Føre-var-prinsippet i matloven
Føre-var-prinsippet er, som tidligere nevnt, lovfestet i norsk rett blant annet i naturmangfoldloven, Svalbardmiljøloven og havressursloven, men i flere andre sammenhenger er dette et ulovfestet prinsipp.
Føre-var-prinsippet er ikke nevnt i selve lovteksten til matloven, men er omtalt en rekke ganger i forarbeidene til loven, Ot.prp. nr. 100 (2002–2003) Om lov om matproduksjon og mattrygghet mv. (matloven). I proposisjonen side 49 heter det:
«Det er en kjensgjerning at det ikke alltid vil være mulig å fremskaffe en vitenskapelig vurdering, fordi det fortsatt kan råde vitenskapelig usikkerhet på området. For å kunne sikre det høye beskyttelsesnivået som er valgt, kan myndighetene i slike tilfeller gjøre bruk av føre-vâr prinsippet og vedta midlertidige risikohåndteringstiltak.»
Videre heter det (s. 51):
«Det er allment anerkjent at vitenskapelige risikovurderinger i visse tilfelle ikke i seg selv kan gi alle de opplysningene som risikohåndteringen bør bygge på. I risikohåndteringen bør det også tas hensyn til andre faktorer som er relevante for spørsmålet som er under vurdering, herunder samfunnsmessige, samfunnsøkonomiske, tradisjonelle, etiske og miljømessige faktorer, samt kontrollmulighetene. I særlige tilfeller der det på bakgrunn av en vurdering av tilgjengelige opplysninger påvises en mulighet for skadelige virkninger på helsen, men hvor det ikke er mulig å gjennomføre en vitenskapelig risikovurdering fordi det råder vitenskapelig usikkerhet, kan lovgiver fastsette midlertidige risikohåndteringstiltak under henvisning til Føre-var-prinsippet i påvente av ytterligere vitenskapelige opplysninger med sikte på en mer omfattende risikovurdering. Føre-var-prinsippet er nødvendig for å kunne sikre gjennomføringen av et høyt beskyttelsesnivå i situasjoner hvor det råder vitenskapelig usikkerhet. Rettslig sett representerer således Føre-var-prinsippet her et nødvendig unntak fra kravet om at regler som har som mål å sikre et høyt beskyttelsesnivå, skal bygge på risikoanalyser. Tiltakene skal stå i forhold til målet, og ikke hindre handelen mer enn nødvendig. Tiltakene skal vurderes på nytt innen rimelig tid, avhengig av arten av den risiko for liv og helse som er påvist, og typen vitenskapelige opplysninger som er nødvendige for å avklare den vitenskapelige usikkerheten og foreta en mer omfattende risikovurdering.»
Selv om føre-var-prinsippet ikke er nevnt eksplisitt i forbindelse med matlovens hjemmel til å forby produkter med gener som koder for antibiotikaresistens (matloven § 9 andre ledd), er det naturlig å se hjemmelen i lys av føre-var-prinsippet. I forarbeidene (s. 77) omtales denne hjemmelen som at den ligger «utenfor» vanlig risikotankegang, som etisk og politisk motivert:
«Hjemmelen for å forby produkter med markørgener som koder for antibiotikaresistens, ble gitt fordi Stortinget ønsket et totalforbud ut fra et så høyt beskyttelsesnivå at det ble vanskelig å innføre totalforbud ut fra vanlig risikotankegang. Det vil fremdeles være behov for å ha en slik hjemmel for etisk og politisk motiverte forbud. Fordi totalforbud er et meget sterkt virkemiddel er det viktig at det er presist og ikke kan misbrukes.»
Føre-var-prinsippet omtales også i forarbeidene i forbindelse med de særskilte merknadene til de ulike bestemmelsene i loven, § 23 om Tilsyn og vedtak. Her er forholdet mellom føre-var-betraktninger og forholdsmessighet uttrykt slik (s. 158):
«Føre-var betraktninger har blitt ansett som et bærende prinsipp i norsk forvaltning, dog uten å være kommet eksplisitt til uttrykk i regelverket. Prinsippet har blant annet vært innfortolket i tilsynsmyndighetens enkeltvedtakskompetanse. Det foreligger forvaltningspraksis som synliggjør at myndighetene blant annet har iverksatt tiltak for å hindre at mulige helseskadelige næringsmidler kommer på markedet, selv om vitenskapelig usikkerhet eller mangel på vitenskapelige data ikke har gjort det mulig å foreta en fullstendig risikovurdering i forkant av beslutningen. Tilsvarende gjelder i forvaltningen av innsatsvarelovgivningen og i forvaltningen av dyre- og plantehelsen. For å videreføre gjeldende forvaltningspraksis vil begrepet «nødvendige vedtak» omfatte vedtak basert på føre- var betraktninger. På grunn av usikkerheten i den konkrete saken, er det ikke gitt at slike vedtak framstår som forholdsmessige. Beslutninger innen offentlig forvaltning forutsettes å være basert på best mulig tilgjengelig kunnskap, og tiltak som iverksettes etter føre- var betraktninger er således ikke prinsipielt forskjellige fra andre forvaltningsvedtak.»
Føre-var-prinsippet og forskrifter til matloven
General Food Law (matlovsforordningen) er siden 2002 det overordnede rammeverket i EØS-området for alt regelverk om mat og fôr. Food Law fastsetter allmenne prinsipper og krav til annet mat- og fôrregelverk i et helkjedeperspektiv og sikrer derved et høyt beskyttelsesnivå med hensyn til helse og forbrukerinteresser, og legger til rette for et effektivt indre marked. Det er også egne bestemmelser om risikoanalyse og om føre-var-prinsippet.
Food Law er gjennomført som en henvisningsforskrift, som betyr at forordningsteksten er tatt inn i sin helhet i forskriften og er gjeldende rett på matområdet. Se nærmere omtale lenger opp.
Forbud mot produkter med antibiotikaresistensgener
Forskrift om forbud mot visse genmodifiserte næringsmidler og § 5 i forskrift om fôrvarer forbyr begge genmodifisert mat og fôr med innhold av funksjonelle antibiotikaresistensgener der disse er tilført ved genmodifisering og kan påvises i sluttproduktet. Forbudene omfatter både GMO omfattet av genteknologiloven, inkludert direktiv-godkjente GMO, og produkter omfattet av matloven.
Dette er et særnorsk, politisk besluttet regelverk, basert på et Stortingsvedtak. Hjemmelsgrunnlaget er matloven § 9 andre ledd som i lovens forarbeider omtales som hjemmelsgrunnlaget for å forby utover vanlig risikotankegang og med mulighet for etiske og politisk motiverte forbud.
Beskyttelsestiltak ved import av ris og risprodukter fra Kina
Forskrift om særskilte beskyttelsestiltak ved import av ris og risprodukter fra Kina er en gjennomføring av tilsvarende regelverk i EU, som er basert på føre-var-prinsippet.
Bakgrunnen for de særskilte beskyttelsestiltakene var mange RASFF-meldinger (Rapid Alert System for Food and Feed) om ulovlig innhold av genmodifisert ris i importerte risprodukter fra Kina, og siden 2008 har EU hatt egne, meget strenge beskyttelsestiltak ved import av ris og risprodukter med opprinnelse fra Kina. I den opprinnelige beslutningen om beskyttelsestiltak, fremkommer det i fortalen nr. 11 at beskyttelsestiltakene tar føre-var-prinsippet med i betraktningen:
«Since the genetically modified rice ‘Bt 63’ is not authorised under Community legislation and in view of the presumption of risk attached to products not authorised according to Regulation (EC) No 1829/2003, which takes into account the precautionary principle laid down in Article 7 of Regulation (EC) No 178/2002, emergency measures should be taken to prevent the contaminated products being placed on the Community market.»
Tiltakene gjelder inntil videre i EØS-området for alle partier av alle produkttyper hvor ris inngår, med krav blant annet om analyserapport og helsesertifikat utstedt av kinesiske myndigheter for hvert eneste vareparti.
Det er et krav i regelverket at beskyttelsestiltakene skal evalueres jevnlig for å vurdere om tiltakene fortsatt er forholdsmessige. EU-kommisjonen innhenter derfor EØS-landenes kontrollresultater for å se om beskyttelsestiltakene virker, og tiltakene evalueres årlig i EUs faste komite for genmodifisert mat og fôr (Standing Committee on Plants, Animals, Food and Feed, Section GM Food and Feed, SCPAFF GMFF). Selv om det de siste årene har vært veldig få påvisninger av ulovlig genmodifisert ris fra Kina, ønsker EU-kommisjonen foreløpig ikke å lempe på tiltakene.
Føre-var-prinsippet og forvaltningspraksis etter matloven
Føre-var-prinsippet gir myndighetene mulighet til å beslutte tiltak for vern om helse eller miljø, selv om de vitenskapelige opplysningene om risikoen ikke gir noe entydig svar, eller på en eller annen måte må anses som ufullstendige. Nøyaktig hvor sterk trusselen eller faren må være før føre-var-prinsippet kan komme til anvendelse, er det vanskelig å fastslå. Rene spekulasjoner vil ikke være nok, men mangel på vitenskapelige data kan alt etter omstendighetene være tilstrekkelig. Under henvisning til føre-var-prinsippet, kan myndighetene treffe midlertidige tiltak om vern av helse eller miljø. Samtidig må myndighetene iverksette tiltak for å få vitenskapelig avkreftet eller bekreftet om den potensielle faren eller trusselen foreligger og om den potensielle faren eller trusselen kan motvirkes med andre, mindre inngripende tiltak.
I tilsynsmyndighetens enkeltvedtakskompetanse kan føre-var-prinsippet spille inn. Begrepet «nødvendige vedtak» omfatter også vedtak basert på føre-var-betraktninger, hvor usikkerhet i en konkret sak kan medføre at enkeltvedtak ikke alltid framstår som forholdsmessige. Beslutninger innen offentlig forvaltning forutsettes likevel alltid å være basert på best mulig tilgjengelig kunnskap, og tiltak som iverksettes etter føre-var-betraktninger er således ikke prinsipielt forskjellige fra andre forvaltningsvedtak.
Ikke-godkjent GMO
Regelverket for godkjenning av genmodifisert mat og fôr i EU og i Norge fastsetter krav om gjennomføring av helse- og miljørisikovurderinger av hhv. EFSA og VKM. Godkjenning gis kun om det ikke påvises noen risiko. Søker må sende inn vitenskapelig dokumentasjon som viser at GMO-en er like trygg for menneskers, dyrs og planters helse og for miljøet som den konvensjonelle varianten den er sammenlignet med.
I lys av risikoanalysen vil føre-var-prinsippet høre hjemme under risikohåndteringen – særlig i situasjoner der en ikke har tilstrekkelig vitenskapelig grunnlag for å kvantifisere sannsynligheten for effekter og konsekvensene av disse.
I EU er det nullaksept for genmodifisert mat og fôr som ikke er godkjent etter unionsregelverket, også om de er helse- og miljørisikovurdert og godkjent i tredjeland. Her ligger føre-var-prinsippet implisitt til grunn, ved at tredjelands risikovurdering ikke oppfattes som tilstrekkelig og at man derfor «påberoper» en grad av vitenskapelig usikkerhet (man mangler kunnskap) som gjør at man iverksetter ulike former for beskyttelsestiltak ved påvisning eller mistanke om ulovlige produkter.
I Norge anerkjennes EFSAs risikovurderinger på lik linje med VKMs vurderinger, og det aksepteres derfor sporforurensninger av EU-godkjente GMO inntil 0,9 % på ingrediensnivå. Men som i EU er det null aksept for innblanding av genmodifisert materiale i mat og fôr som er godkjent i tredjeland og ikke i EU. Genmodifisert mat og fôr som ikke er godkjent, har man ikke kunnskap om når det gjelder mulig helse- og miljørisiko. Føre-var-prinsippet ligger med andre ord også her til grunn som en del av risikohåndteringen.
Tilsyn og kontroll
Ingen genmodifiserte mat- og fôrprodukter er godkjent i Norge per i dag. Mattilsynets overvåkning og kontroll av markedet for ulovlig innhold av genmodifisert materiale kan sees på som et føre-var-tiltak. Ikke-godkjent genmodifisert mat og fôr av ukjent opprinnelse har norske myndigheter ingen kunnskap om, og føre-var-prinsippet gjør at virksomhetene er pålagt rutiner for å unngå slike ulovlige produkter. Mattilsynet gir pålegg om omsetningsforbud hvis det påvises ulovlige produkter ved analyse.
6.7.5 Føre-var-prinsippet i lys av generelle WTO-forpliktelser
WTO-avtalene gir medlemslandene rettigheter og forpliktelser når det gjelder anvendelse av føre-var-prinsippet. Det er mulig å innføre midlertidige føre-var-tiltak i de tilfellene der det foreligger en potensiell risiko, men hvor det ikke finnes god nok kunnskap til å dokumentere risikoen. Tiltaket må imidlertid framstå som proporsjonalt og ikke-diskriminerende. I tillegg er det en forpliktelse om at man må opptre aktivt for å søke å fremskaffe nødvendig vitenskapelig kunnskap innen rimelig tid. WTOs appell-domstol har klargjort kriterier for bruk og opprettholdelse av føre-var-tiltak, se for eksempel omtalen av Bioteknologisaken (EC – Measures Affecting the Approval and Marketing of Biotech Products)47 omtalt i kapittel 5.4.4.
6.8 Saksgang i Norge for nasjonale søknader og søknader til EU
6.8.1 Forholdet til EUs GMO-regelverk
Norge er etter EØS-avtalen forpliktet til å følge EU-reglene som gjelder blant annet for mattrygghet og matproduksjon, og for miljøområdet med blant annet direktiv 2001/18/EF (utsettingsdirektivet). Rettsaktene på matområdet er i volum det største rettsområdet i EØS-avtalen og utgjør anslagsvis 40 % av rettsaktene innlemmet i EØS-avtalen. Disse er gjennomført i matloven med General Food Law (matlovsforordningen) som overordnet rammeverk, med unntak av regelverket om genmodifisert mat og fôr (den såkalte GM-pakken). Utsettingsdirektivet er en del av EUs miljøregelverk og er gjennomført i genteknologiloven.
6.8.2 Søknader til Norge
Genmodifisert mat og fôr som er godkjent i EU, er ikke lovlig å omsette i Norge da Norge ikke har innlemmet EUs regelverk på dette området i EØS-avtalen per i dag. Virksomheter som ønsker å selge slike produkter i Norge må sende søknad til Mattilsynet for prosessert GMO til bruk som mat og fôr, eller til Miljødirektoratet for levende GMO. Dersom det søkes om tillatelse til anvendelse av den samme GMO-en i både prosessert og levende tilstand, søkes begge myndigheter. Per mai 2023 er det ingen nasjonalt godkjente GMO mat- og fôrprodukter på det norske markedet.
Norge har mottatt og behandlet nasjonale søknader under genteknologiloven om innesluttet bruk, og om klinisk utprøving av GMO-legemidler til mennesker. Det har imidlertid ikke kommet nasjonale søknader om klinisk utprøving av GMO-legemidler til dyr eller om utsetting av levende GMO til omsetning.
Frem til sommeren 2022 var det ikke kommet noen søknader om godkjenning av genmodifisert mat og fôr etter matloven. Da mottok Mattilsynet den første norske søknaden om godkjenning av rapsolje fra en genmodifisert raps med øket innhold av langkjedete omega-3 fettsyrer. Omsøkt bruksområde i Norge er kun som ingrediens i fiskefôr. VKM har foretatt en risikovurdering av oljen48, som vil være basis for Mattilsynets vurdering og senere vedtak i saken. VKM bruker de samme retningslinjene for risikovurdering som EFSA, se nærmere omtale av dette i kapittel 8.
6.8.3 Søknader til EU
EUs regelverk om genmodifisert mat og fôr består av to basisrettsakter som hjemler krav om godkjenning, merking og sporbarhet. I dette regelverket skilles det ikke mellom levende, formeringsdyktige GMO og prosesserte/bearbeidede produkter fra GMO. Også produkter uten DNA og proteiner fra genmodifiseringen er omfattet av regelverket. Regelverket omfatter alle GMO som forventes å kunne inngå i mat- eller fôrkjeden uavhengig av om de er levende eller døde og uavhengig av om det er en tilsiktet eller utilsiktet innblanding av genmodifisert materiale. Regelverket hjemler strenge krav til helserisikovurdering og krav til håndtering av helserisiko. Det refereres til utsettingsdirektivet for krav om miljørisikovurdering og krav til håndtering av miljørisiko.
Søknader til EU om godkjenning av levende GMO som ikke forventes å inngå i mat og fôrkjeden, behandles etter utsettingsdirektivet. De senere år er det kun søknader om import og omsetning av ulike avskårne GMO nellikvarianter som prydblomster som er godkjent under direktivet i EØS-området (dvs. inkludert Norge). Utsettingsdirektivet utgjør en av basisrettaktene også på GMO mat- og fôrområdet i EU, fordi det omfatter GMO-definisjonen samt kravene til miljørisikovurdering av GMO som søkes godkjent etter GM mat- og fôrforordningen (forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr), og miljøovervåkingsplan. Utsettingsdirektivet i EU hjemler altså ikke lenger godkjenningskrav for levende GMO mat og fôr, men innehar GMO-definisjonen og kravene til miljørisikovurdering og miljøovervåkingsplan for alle GMO. Genteknologilovens virkeområde er ikke endret tilsvarende i Norge.
6.8.4 Nasjonal forbehandling av søknader under GM mat- og fôrforordningen
Norge har ingen plikt til å realitetsbehandle søknader som sendes til EU under denne forordningen, siden denne ikke er innlemmet i EØS-avtalen. Men hvis man legges Norges utkast til tilpasningstekst til grunn (basert på tilpasningsteksten til utsettingsdirektivet), er det viktig at søknadene vurderes parallelt med EU slik at man rekker å enten stadfeste godkjenningen eller sluttføre en prosedyre for å legge ned norsk forbud mot søknaden innen 30-dagers fristen som foreslås i utkastet.
Norge har, på lik linje med medlemsstatene i EU, tilgang til alle søknader med dokumentasjon som sendes til EU under GM mat- og fôrforordningen. I EU sendes ikke selve søknaden på en offentlig høring, i stedet sender EFSA ut den ferdige risikovurderingen på en måneds offentlig høring. EFSA sender imidlertid ut søknadene med søkers dokumentasjon på myndighetshøring i medlemsstatene og i EFTA-landene, og Norge kan gi vitenskapelige innspill til denne høringen på lik linje med medlemsstatene. Norge deltar også på lik linje med medlemsstatene i vurdering av søknaden frem til votering i komitéen som behandler søknadene (SCPAFF GMFF) hvor Norge «går på gangen». Det er derfor viktig at Norge gir innspill på riktig tidspunkt i søknadsprosessen. I de tilfeller der Norge kan ha innsigelser til en mulig godkjenning i EU, er det spesielt viktig å legge frem disse i SCPAFF GMFF når EFSA er tilstede og legger frem risikovurderingene med innspillene fra medlemslandene.
Det viktigste forholdet i godkjenningsprosessen i EU er en helse- og miljørisikovurdering av aktuell GMO for å sikre at det ikke forekommer helserisiko for mennesker eller dyr, eller uheldige konsekvenser for miljøet. EFSA foretar disse risikovurderingene i EU.
Mattilsynet og Miljødirektoratet benytter VKM til å vurdere GMO-ene som søkes godkjent i EU, men etatene vil foreta den endelige vurderingen av de enkelte produktene. I 2020 fikk VKM et nytt felles oppdrag fra Mattilsynet og Miljødirektoratet om å vurdere GMO til mat og fôr som søkes godkjent i EU. Oppdraget er revidert i 2023. Hensikten med oppdraget er å forkorte og forenkle prosessen i VKMs faggruppe for GMO.
VKM skal vurdere dokumentasjonen som følger søknader om godkjenning av GMO i EU og eventuelt spille inn til EFSAs vitenskapelige høring av søknaden. VKM skal deretter vurdere om det er forhold som kan indikere en særnorsk helse- eller miljørisiko sammenlignet med i EU. Dersom VKM finner særnorske forhold, skal VKM utføre en full risikovurdering for å belyse disse forholdene. Når VKM ikke finner særnorsk risiko, vil EFSAs risikovurdering være dekkende også for norske forhold. Norske myndigheter anerkjenner EFSAs risikovurderinger.
VKM vurderer først søkers dokumentasjon og gir innspill til EFSAs vitenskapelige høring med kopi til Mattilsynet og Miljødirektoratet. Neste trinn i vurderingen er å sjekke ut om søker har fulgt kravene til søknaden som fremgår av forordning (EU) nr. 503/2013 om søknader om godkjenning av GMO mat og fôr49, utsettingsdirektivet og EFSAs egne retningslinjer, vurdere annen relevant vitenskapelig litteratur, samt å vurdere om det er særnorsk helse- og/eller miljørisiko som i så fall vil medføre at VKM må gjøre en full helse- og miljørisikovurdering. VKM skal også påpeke på hvilken måte eller på hvilket område det er kunnskapshull, og det skal fremgå om denne kunnskapsmangelen påvirker konklusjonen i vurderingen.
VKMs vurderinger skal sikre at Norge har et godt nok grunnlag på et riktig tidspunkt, til å følge opp søknadsprosessen i EU med spørsmål, innsigelser og informasjon om eventuelle særnorske forhold som kan gi annen helse- eller miljørisiko i Norge. Vurderingene skal også sikre at Norge har tilstrekkelig med tid til å ta endelig stilling til en søknad kort tid etter at den er godkjent i EU. Etter innlemmelse av GM-pakken, vil dette være særlig viktig i de tilfeller hvor Norge vurderer å forby en GMO til mat og fôr i stedet for å gjenta EUs beslutning om godkjenning.
6.8.5 Søknader om GMO til utsetting i miljøet etter utsettingsdirektivet
Søknader om forsøksutsetting og annen utsetting
Miljødirektoratet er vedtaksmyndighet for forsøksutsetting og utsetting i veksthus, akvakulturinnretning, dyrestaller o.l. (som ikke er godkjent for innesluttet bruk), mens Klima- og miljødepartementet er vedtaksmyndighet for annen utsetting av levende GMO, inkludert omsetning. VKM vurderer på oppdrag fra Miljødirektoratet opplysninger og dokumentasjon i søknaden, og risikovurdering til søker. Bioteknologirådet kan bes om innspill til bærekraft, samfunnsnytte og etikk av søknaden. Miljødirektoratet lyser søknaden ut på høring, og fatter endelig beslutning om søknaden basert på vurderingene til VKM og Bioteknologirådet, og eventuelle innkomne høringsinnspill.
Søknader om omsetning (jamfør felles EU-prosedyre etter utsettingsdirektivet)
For søknader om omsetning av levende GMO til annet enn mat og fôr, er Norge knyttet til felles prosedyre i EU gjennom utsettingsdirektivet. Klima- og miljødepartementet har 5. juli 2017 fastsatt saksbehandlingsprosedyrer for slike søknader. Prosedyrene kommer til anvendelse både der søknaden fremmes i et annet EØS-land og hvis søknaden fremmes i Norge. Rutinene forenkler tidligere rutiner og legger til rette for at Norge kan komme til en avgjørelse kort tid etter godkjenning av en GMO i EU.
Norge deltar på lik linje med medlemslandene i EUs saksbehandling og kan gi innspill underveis i søknadsbehandlingen, men kan ikke votere over søknaden. Miljødirektoratet koordinerer slike søknader i Norge og opp mot EU, og leverer endelig tilrådning om norske vedtak til Klima- og miljødepartementet.
Dersom Norge er mottakerland for søknaden, skal Miljødirektoratet utarbeide en tidsplan for behandlingen av søknaden, og snarest informere berørte organer. VKM vil etter rutinene bli bedt om å vurdere opplysninger og dokumentasjon i søknaden, og utarbeide en risikovurdering av den omsøkte GMO-en. Mattilsynet vil bli bedt om å gi en vurdering av søknaden innenfor sine ansvarsområder til Miljødirektoratet. Bioteknologirådet vil bli bedt om å innhente og vurdere informasjon vedrørende bærekraft, samfunnsnytte og etikk. Vurderingsrapporten med tilrådning sendes av Miljødirektoratet sammen med søknaden til Kommisjonen etter konsultasjon med KLD, og behandles videre i EU-prosedyren med to innspillsrunder for EU/EØS-landene om søknaden.
I motsetning til under forordning (EF) nr. 1829/2003 om genmodifisert mat og fôr, så er det Kommisjonen som koordinerer søknadsbehandlingen i EU for søknader etter utsettingsdirektivet, som består av to høringsrunder av EU/EØS-landene. Det er mottakslandet for søknaden som forelegger første risikovurdering av søknaden til behandling. Dersom landene har innsigelser til risikovurderingen, søkers opplysninger eller annet etter de to høringsrundene av landene, sendes søknaden til risikovurdering av EFSA.
Ved godkjenning av søknaden i EU, er søknaden automatisk godkjent i Norge med mindre norske myndigheter legger ned særnorsk forbud mot søknaden. Slike forbud fattes av regjeringen (Kongen i statsråd).
Helse- og miljørisikovurdering
En sentral del av konsekvensutredningen er vurdering av eventuelle konsekvenser av den genmodifiserte organismen i miljøet, og menneskers og dyrs helse. VKM utfører denne vurderingen på samme måte som for forordningssøknadene til EU, se kapittel 6.8.4. Miljørisikovurderingen bør utføres med sikte på å fastslå hvorvidt det er behov for risikostyring, og i så fall, hvilke metoder som vil være mest hensiktsmessige. VKM, som uavhengig vitenskapelig organ, utfører risikovurdering av søknader om GMO etter utsettingsdirektivet. Miljødirektoratet er ansvarlig for miljørisikohåndtering og oppdrag til VKM om miljørisiko, mens Mattilsynet er ansvarlig for helserisikohåndtering og oppdrag til VKM om helserisiko. Etatene har et felles oppdrag til VKM for søknader mottatt etter henholdsvis utsettingsdirektivet og GM mat- og fôrforordningen. VKMs vurderinger skal sikre at Norge har et godt nok grunnlag på et riktig tidspunkt, til å følge opp søknadsprosessen i EU med spørsmål, innsigelser og informasjon om eventuelle særnorske forhold. Vurderingen inkluderer vitenskapelig vurdering av opplysninger og dokumentasjon fra søker, og utforming av spørsmål i forbindelse med EU-behandlingen av søknaden. Dersom det er innsigelser til søknaden og/eller særnorske forhold ved søknaden som er identifisert skal VKM utføre en full risikovurdering for GMO-en.
Bærekraft, samfunnsnytte og etikk (BSE)
Bioteknologirådet er ansvarlig for vurdering av den enkelte søknad opp mot kriteriene bærekraft, samfunnsnytte og etikk under genteknologiloven. Det innebærer innhenting av informasjon og utarbeidelse av kunnskapsgrunnlag basert på publisert faglig relevant litteratur og annen relevant informasjon for vurderingen av GMO-ens innvirkning på bærekraft, samfunnsnytte og etikk. VKMs og EFSAs vurderinger skal legges til grunn for rådets vurderinger, hva gjelder vurderinger som bygger på helse- og miljømessige konsekvenser av GMO-en. Bioteknologirådet kan også ha direkte kontakt med søker vedrørende spørsmål om bærekraft, samfunnsnytte og etikk.
Det er mulig å fremme spørsmål om sosio-økonomiske effekter av GMO-er fremmet etter utsettingsdirektivet. Miljødirektoratet spiller derfor eventuelle spørsmål om bærekraft, samfunnsnytte og etikk inn i EUs innspillsrunder for medlemslandene, og søker kan svare på disse gjennom EU-prosedyren.
6.8.6 Søknader om GMO til innesluttet bruk
Norge er tilsluttet EUs direktiv om innesluttet bruk av genmodifiserte mikroorganismer. Det meste av innesluttet bruk regelverk for planter og dyr m.m er nasjonalt regelverk og forvaltes av Helse- og omsorgsdepartementet og Helsedirektoratet, og omtales ikke nærmere her.
6.9 Koblinger mellom GMO mat- og miljøregelverket i EU
6.9.1 Innledning
Nesten hele matområdet og det meste av miljøområdet, herunder GMO, er omfattet av EØS-avtalen. Det betyr at Norge i utgangspunktet er forpliktet til å innlemme EUs rettsakter på disse områdene. Det er derfor viktig å se på koblinger i EUs regelverk samt vurdere eventuelle handlingsrom vi har i Norge samt mulighetene vi har for å få tilpasninger.
Før EU fikk et samlet regelverk for genmodifisert mat og fôr i 2003, var det ulike regelverk som regulerte ulike bruksområder for den samme GMO og ingen felles regler verken for godkjenning, merking eller sporbarhet.
Direktiv 90/220/EØF for utsetting av genmodifiserte organismer i miljøet var det første regelverket som regulerte godkjenningskrav for utsetting av levende GMO til mat, fôr, legemidler og annen bruk med basis i miljørisiko. I 1997 fastsatte EU et eget regelverk for matvarer utviklet ved bruk av nye teknologier, herunder GMO. Med Ny mat-regelverket (forordning (EF) nr. 258/97) ble levende GMO til mat tatt ut av utsettingsdirektivet, og både levende og prosessert (bearbeidet/avledet) GMO til bruk som mat ble nå godkjent i hht. Ny mat-forordningen. Utsettingsdirektivet regulerte etter dette levende GMO til annen bruk enn mat, og det var ingen godkjenningskrav for prosessert GMO til fôr. På denne tiden fantes det heller ikke et sentralt risikovurderingsorgan.
Godkjenningsprosedyren under utsettingsdirektivet og Ny mat-forordningen var todelt. Det enkelte land mottok en søknad om godkjenning av en GMO med en påfølgende høring av andre medlemsstater. Hvis ingen hadde innsigelser til en nasjonal godkjenning eller forbud, hadde godkjenningen eller forbudet virkning for hele unionen. Hvis et land derimot hadde innvendinger mot forslaget om forbud eller godkjenning, gikk søknaden gjennom en voteringsprosess i ekspertgruppene under EU kommisjonen.
Fra 1999 nektet flere medlemsstater generelt å godkjenne GMO til bruk som mat og fôr før man fikk på plass nye fellesskapsregler for helserisikovurdering, merking og sporbarhet. Perioden frem til nytt regelverk om genmodifisert mat og fôr ble vedtatt høsten 2003 (GM-pakken), kalles moratorium-perioden fordi ingen GMO ble godkjent i denne perioden i EU. Det førte til at USA gikk til WTO-panelsak, som de i hovedsak vant, fordi de mente EU hindret handel i strid med WTO-regelverket. Panelsaken medvirket sterkt til at det nye regelverket for genmodifisert mat og fôr ble laget. Hovedhensikten var å sette et høyt beskyttelsesnivå med basis i vitenskapelige risikovurderinger og med fokus på forbrukerrettigheter – samtidig som regelverket ikke skulle medføre handelshindringer og brudd på WTO-avtalen.
6.9.2 Nærmere om EUs forvaltning av GMO-regelverket
Genteknologiloven ble vedtatt av Stortinget i 1993 og gjennomførte EUs første utsettingsdirektiv (90/220/EØF) og senere EUs andre utsettingsdirektiv (2001/18/EF). Med Ny mat-regelverket i EU ble levende GMO til mat tatt ut av utsettingsdirektivet, og med EUs nye fellesskapsregelverk om genmodifisert mat og fôr på plass (GM-pakken), var heller ikke levende GMO til bruk som fôr omfattet av utsettingsdirektivet. GM mat- og fôr regelverket i EU regulerer altså levende og død mat under ett regelverk, men viser som tidligere nevnt til utsettingsdirektivet for krav om miljørisikovurdering, GMO-definisjonen og miljøovervåkningsplan.
En rekke ulike faktorer kan ha en direkte eller indirekte innvirkning på mattryggheten, og Food Law’s virkeområde dekker hele matområdet, herunder også godkjenning mv. av GMO mat og fôr. EUs matregelverk går helt fra primærproduksjonen av mat og fôr og helt frem til overlevering av maten til forbrukerne – «from farm to fork». Innenfor primærproduksjon vil blant annet dyrehelse, såvarer, gjødsel og plantevernmidler være omfattet, og forordningen berører også spørsmål knyttet til dyrevelferd, miljøvennlig produksjon og dyre- og plantehelse, i tillegg til alle ledd i produksjon av mat og fôr frem til ferdig produkt omsettes inkludert helse- og miljørisikovurderinger, merking, emballasje, markedsføring, toll m.fl.
EUs generelle, betydelige fokus på bærekraft, inkludert i Farm to fork-strategien, vil gi en tilleggsdimensjon i form av et nytt, overordnet rammeverk for å sikre bærekraftige matsystemer. Forventede nye krav til bærekraft vil da gjelde matområdet generelt, og især produktgrupper som er omfattet av godkjenningsordninger slik som genmodifisert mat og fôr (både levende og prosessert). I Norge er hensyn til bærekraft lovfestet i genteknologiloven (levende GMO til for eksempel mat og fôr), men ikke på tilsvarende måte i matloven (prosessert, ikke-formeringsdyktig GMO til mat og fôr).
Rettsaktene i GM-pakken og kontrollforordningen er selve rammeverket for godkjenningskrav, forbrukerrettigheter og sporbarhet, kontroll og overvåkning for GMO til bruk som mat og fôr. Her hjemles reglene for blant annet helserisikovurderingen, godkjennings-, merke- og sporbarhetskrav og myndighetskontrollen. Regelverket henviser til utsettingsdirektivet for definisjonen av GMO og for krav til miljørisikovurderingen av GMO samt miljøovervåkningsplan.
Som tidligere nevnt, er utsettingsdirektivet ikke hjemlet i General Food Law, men likevel omfattet av kontrollforordningen fordi GMO godkjent til annen bruk enn mat og fôr kan komme inn i matkjeden. Av samme grunn er utsettingsdirektivet endret flere ganger for å ta opp i seg ulike krav gitt i GM-pakken, for eksempel et felles identifikatorsystem for GMO som godkjennes til mat, fôr eller annen bruk, samt sporbarhetskrav for levende GMO. Utsettingsdirektivet er også endret som følge av nye åpenhetskrav i åpenhetsforordningen (forordning (EU) nr. 2019/1381). Det totale rammeverket for genmodifisert mat og fôr inkluderer derfor utsettingsdirektivet. EU-kommisjonen gjennomførte et skille i GMO-forvaltningen i 2009, da miljøforvaltningen av GMO, også utsettingsdirektivet, ble overført organisatorisk fra generalenheten DG ENVI i EU-kommisjonen til DG SANTE – altså fra «miljøforvaltningen» til «mattrygghets- og forbrukerforvaltningen.»
EFSA er EUs sentrale risikovurderingsorgan, både med hensyn til gjennomføring av risikovurderinger på ulike områder (blant annet GMO etter både forordningen og direktivet), men også med hensyn til å utvikle retningslinjer for risikoanalysen. Kravene til helserisikovurderinger under GMO-regelverket finnes i en egen forordning supplert av flere retningslinjer EFSA har laget. Kravene til miljørisikovurdering finnes i utsettingsdirektivet og er også supplert av flere retningslinjer laget av EFSA.
Regelverket om risikovurderingsprosessen og vurderings- og godkjenningsprosessen i EU har med GM-pakken blitt meget omfattende. Det har likevel bare én inngangsdør for søker og med en enhetlig og helhetlig søknadsbehandling hvor vitenskapelige risikovurderinger av et eget risikovurderingsorgan, regler for bruk av føre-var-prinsippet og sterke forbrukerrettigheter skal sikre et meget høyt beskyttelsesnivå med hensyn til helse, miljø og forbrukere.
6.9.3 Oppsummert om konkrete koblinger mellom GMO-regelverk i EU
Utsettingsdirektivet definerer ‘genmodifisert organisme’. Annet GMO-regelverk henviser til denne definisjonen, og direktivet regnes derfor som en av basisrettsaktene også når det gjelder genmodifisert mat og fôr, selv om det ikke regnes som en del av GM-pakken.
GM mat- og fôrforordningen regulerer godkjennings- og merkekrav både til GMO og avledete produkter, med basis i krav om helse- og miljørisikovurdering. For miljørisikovurderingen refereres det da til utsettingsdirektivet, mens krav om helserisikovurdering finnes i utfyllende regelverk til GM-pakken samt i ulike veiledningsdokumenter fra EFSA. En søknad om godkjenning kan omfatte all bruk, dvs. dyrking, import, bruk som mat og fôr og annen bruk enn mat og fôr (for eksempel import av råvare for prosessering til emballasje, papir og råmateriale til tekstiler). En søknad kan også omfatte all bruk unntatt dyrking, og i praksis i dag får EU kun søknader med dette omfanget. Det er imidlertid ikke mulig å søke om godkjenning for et smalere bruksområde enn dette, for eksempel kun dyrking av GMO såvare til mat og fôr eller kun bruk som enten prosessert eller levende GMO. Årsaken er at alle bruksområder skal helse- og miljørisikovurderes slik at man vet at det ikke foreligger risiko ved utilsiktet bruk av GMO-en. En slik søknad om godkjenning kan imidlertid fremmes etter utsettingsdirektivet, men antas å ha begrenset verdi da GMO-avlingen eller GMO-dyret ikke kan brukes direkte som mat og fôr og heller ikke kan prosesseres til andre produkter uten etter godkjenning under GM mat- og fôrforordningen. Det er likevel mulig å eksportere avlingen eller GMO-dyret til land utenfor EØS, men i praksis sendes alle søknader om godkjenning av GMO til bruk som mat og fôr, under forordningen.
Sporbarhets- og merkeforordningen er hjemlet i Food Law, men den endret utsettingsdirektivet ved å endre sporbarhets- og merkekrav slik at disse på flere punkter for GMO som ikke er mat og fôr, settes tilsvarende som for GMO som er mat og fôr. Sporbarhetskravet er et viktig element for å kunne trekke tilbake en GMO fra matkjedene ved nye opplysninger om at produktet medfører helse- eller miljørisiko.
Åpenhetsforordningen gjelder for ulike typer matregelverk under Food Law (syv forordninger inkludert GM mat- og fôrforordningen), men gjelder også for utsettingsdirektivet som ikke er hjemlet i Food Law. Dette grepet har EU-kommisjonen valgt på grunn av koblingen opp mot GM mat- og fôrregelverket, hvor miljørisikovurderingene gjøres etter krav i utsettingsdirektivet.
6.10 Koblinger mellom GMO mat- og miljøregelverket i Norge
6.10.1 Status implementering av GM-pakken
Regelverket om genmodifisert mat og fôr trådte i kraft i EU i 2004. Etter vanlige prosedyrer og de forpliktelser Norge har etter EØS-avtalen skulle GM-pakken vært innlemmet og gjennomført kort tid etter EU (såkalt rettidig gjennomføring). EUs regelverkspakke om genmodifisert mat og fôr fra 2003 er EØS-relevant, og EFTA-landene er i forhandlinger med EU om tilpasningstekst. Det er usikkert når GM-pakken kan innlemmes i EØS-avtalen.
I Norge har myndighetene hatt behov for tid til å vurdere betydningen av innlemmelse, da visse elementer i regelverket er ansett som problematiske. Det gjelder særlig den sentraliserte godkjenningsprosedyren som følger av GM mat- og fôrforordningen med basis i EFSAs vurdering av helse- og miljørisikovurdering og påfølgende votering hvor Norge ikke har rett til å avgi stemme. Videre har man vurdert hvilke mulighetsrom Norge har til å eventuelt forby genmodifiserte produkter. Det er også ansett som viktig å kunne opprettholde genteknologilovens tre vurderingskriterier om etikk, bærekraft og samfunnsnytte i vurderingen av levende GMO.
Tidligere regjeringer har funnet regelverket EØS-relevant, og det er laget utkast til tilpasningstekst for innlemmelse av GM-pakken i EØS-avtalen. Den foreslåtte tilpasningsteksten er basert på tilpasningsteksten til utsettingsdirektivet. Denne gir adgang til å fatte avvikende, nasjonale beslutninger sammenlignet med EU, og opprettholder genteknologilovens krav til etikk, samfunnsnytte og en bærekraftig utvikling.
Regelverksteknisk kan en implementering av GM-pakken i Norge gjøres på ulike måter og gjennomføres under ett eller flere regelverk. Dette må avklares i forbindelse med videre prosess.
6.10.2 Dagens rettstilstand
Genteknologiloven fra 1993 gjennomførte EUs første utsettingsdirektiv (90/220/EØF). På den tiden var det ikke utviklet et eget felles regelverk i EU om godkjenning av genmodifisert mat og fôr. All omsetning av levende GMO ble regulert av utsettingsdirektivet i EU og av genteknologiloven i Norge, og regelverkene for omsetning skilte ikke mellom de forskjellige bruksområdene. I 1997 fastsatte EU et eget regelverk for matvarer utviklet ved bruk av nye teknologier, herunder GMO. Med Ny mat regelverket i EU ble alle genmodifiserte organismer til mat tatt ut av utsettingsdirektivet, og senere overført til GM mat- og fôrforordningen. Genmodifiserte organismer til fôr ble likeledes overført fra utsettingsdirektivet til forordningen i 2003. Tilsvarende endringer ble ikke gjort nasjonalt i Norge. GM mat- og fôrforordningen er ikke innlemmet i EØS-avtalen.
Forvaltningen av regelverk om genmodifisert mat og fôr i Norge er i dag delt mellom mat- og miljøforvaltningen. GMO-regelverket i Norge er både EØS-regelverk og nasjonalt regelverk. Genteknologiloven regulerer godkjenning av levende GMO, både til mat og fôr og til annen bruk. Det kreves ikke godkjenning for omsetning av en GMO som er godkjent for omsetning i et annet EØS-land under utsettingsdirektivet. For GMO-er som ikke er godkjent, kreves nasjonale søknader om godkjenning.
Matloven regulerer i hovedsak ikke-formeringsdyktig mat og fôr laget fra GMO (prosesserte/bearbeidede produkter). Forskrifter under matloven regulerer likevel merking både av levende og bearbeidet GMO til mat og fôr, godkjenning og merking av genmodifiserte såvaresorter samt visse typer forbud mot levende og bearbeidet GMO. Regelverket er nasjonalt i påvente av innlemming av GM-pakken i norsk rett. Regelverk for grensekontroll av ris og risprodukter fra Kina er EØS-regelverk, gjennomført i kontrollforordningen.
Genmodifisert mat og fôr godkjent i EU under GM mat- og fôrforordningen er ikke godkjent for omsetning i Norge. En virksomhet som ønsker godkjenning av både levende og prosessert genmodifisert mat og fôr i Norge, må derfor sende separat søknad om godkjenning av levende GMO mat og fôr til Miljødirektoratet, og separat søknad om godkjenning av bearbeidede produkter fra den samme GMO-en til Mattilsynet.
6.10.3 Nærmere om norsk forvaltning av GMO-regelverket
Det viktigste målet i norsk matpolitikk er å sikre helsemessig trygg mat til forbrukerne i Norge og i de markeder vi eksporterer til. I tillegg er det et helt sentralt mål at maten oppleves som trygg. I dette ligger det at myndighetene skal ha et spesielt fokus på forbrukernes behov for informasjon og kunnskap. Videre må ikke utsetting av godkjent GMO medføre miljørisiko, og miljøvennlig produksjon skal ivaretas. Godkjenningsordningen er basert på en grundig helserisikovurdering etter de samme prinsipper som i EUs regelverk om genmodifisert mat og fôr, og en grundig miljørisikovurdering etter de samme prinsipper som i EUs utsettingsdirektiv. I tillegg vurderes levende GMO etter de nasjonale kravene om bærekraft, samfunnsnytte og etikk. De nærmere vurderingene og vektleggingen av de sistnevnte kriteriene kan variere, slik erfaringene i Norge med søknader etter utsettingsdirektivet viser.
Virksomheter som ønsker å omsette genmodifisert prosessert mat og fôr, må sende søknad til Mattilsynet som vil bruke VKM til å gjennomføre en risikovurdering. Mattilsynet har fått delegert vedtaksmyndighet fra matdepartementene til å behandle søknader om godkjenning av prosessert genmodifisert mat og fôr og til å vedta godkjenning eller forbud.
Etter en eventuell godkjenning, må alle produkter med innhold av GMO-en merkes etter egne krav, uavhengig om det kan påvises DNA eller protein som stammer fra genmodifiseringen (f.eks. rapsolje fra GMO rapsfrø, sukker fra GMO sukkerbete). Det er ingen godkjente, genmodifiserte prosesserte mat- og fôrprodukter på markedet etter matloven i dag, men Mattilsynet behandler våren 2023 den første søknaden til Norge om godkjenning av olje produsert fra genmodifisert raps (se kapittel 6.4.3.3). Nasjonal godkjenningsordning under matloven vil gjelde fram til Norge implementerer EUs regelverk om genmodifisert mat og fôr.
Det er heller ingen godkjente levende GMO til mat og fôr i Norge, og det er heller ingen som har sendt inn søknad til Norge (dvs. Miljødirektoratet). Godkjenningsordningen etter genteknologiloven er basert på en grundig helserisikovurdering (der koordineringsansvar ligger hos Mattilsynet) etter de samme prinsipper som i EUs regelverk om genmodifisert mat og fôr, og en grundig miljørisikovurdering (Miljødirektoratet har koordineringsansvaret) etter prinsippene i genteknologiloven og utsettingsdirektivet (EØS-regelverk). Virksomheter som ønsker å omsette levende GMO mat og fôr med eller uten dyrking i Norge, må sende søknad til Miljødirektoratet som vil bruke VKM til å gjennomføre en miljørisikovurdering. Mattilsynet ber VKM om en helserisikovurdering og en vurdering av agronomisk miljørisiko og sender en overordnet helhetsvurdering av disse forholdene til Miljødirektoratet sammen med en vurdering av sameksistenstiltak hvis søknaden omfatter dyrking. Miljødirektoratet ber Bioteknologirådet om å komme med råd om bærekraft, samfunnsnytte og etikk og foretar til slutt en helhetlig vurdering og anbefaling som sendes til KLD som fatter vedtak om avslag eller godkjenning. Etter en eventuell godkjenning av GMO-en, må den merkes etter krav i matloven før den kan omsettes (for eksempel mais til dyrking, import av rapsfrø til pressing). Gjelder søknaden dyrking i Norge, kan ikke dyrkingen tillates før det er søkt om sortsgodkjenning under matloven og Mattilsynet har fastsatt vilkår for dyrkingen i eget enkeltvedtak.
I Norge anerkjennes EFSAs risikovurderinger av genmodifisert mat og fôr på lik linje med VKMs vurderinger. Det er derfor ikke godkjenningskrav for sporforurensninger av EU-godkjent GMO-materiale inntil 0,9 % på ingrediensnivå under visse forutsetninger. Dette er regelverksfestet under matloven, og etablert praksis under genteknologiloven med unntak av for såvarer hvor det er en nullgrense. Som i EU er det for øvrig ingen aksept for innblanding av genmodifisert materiale i mat og fôr som ikke er godkjent, selv om de eventuelt er godkjent i tredjeland. Begrunnelsen er at genmodifisert mat og fôr som ikke er godkjent i EU, har man ikke kunnskap om når det gjelder mulig helse- og miljørisiko. Føre-var-prinsippet ligger med andre ord til grunn som en del av risikohåndteringen.
Siden søknader om genmodifisert mat og fôr behandles under GM mat og fôr-regelverket i EU, som ikke er innlemmet i norsk rett, gjør ikke Norge en fullbehandling av søknadene til EU. Norge (Mattilsynet og Miljødirektoratet) deltar dog allerede i dag i ekspertkomiteen for genmodifisert mat og fôr, og VKM gir innspill på vegne av myndighetene til EFSAs vitenskapelige høring av søknadene. VKM gjennomfører dessuten vurderinger av alle GMO som søkes godkjent i EU med henblikk på om det er annen helse- og miljørisiko for GMO-en i Norge. Finner VKM at det kan være særnorsk risiko, vil Mattilsynet ta opp dette i ekspertkomiteen når EFSA gjennomgår sin endelige risikovurdering.
6.10.4 Vedtak etter dagens organisering av forvaltningen – nasjonale søknader
I EU vil den enkelte GMO som godkjennes til bruk som mat og fôr, i regelen være tillatt for alle bruksområder (one door-one key). I Norge vil det for søknader som sendes til Norge etter dagens regulering, og som omfatter både levende og prosessert GMO, fattes to vedtak for den samme GMO-en – bruksområde prosessert mat og fôr etter matloven og bruksområde levende mat og fôr under genteknologiloven (two doors-two keys).
Avhengig av om det påvises forhold i risikovurderingen og/eller om det påvises positive eller negative konsekvenser etter kriteriene etikk, bærekraft og samfunnsnytte, kan blant annet følgende hovedalternativer for vedtak være mulige:
Ikke påvist helse- eller miljørisiko eller negative konsekvenser med hensyn til de tre tilleggskriteriene vurdert for levende GMO: Godkjenning av bearbeidede mat- og fôrprodukter etter matloven, og godkjenning av den samme GMO til utsetting etter genteknologiloven.
Ikke påvist helse- eller miljørisiko, men negative konsekvenser med hensyn til tilleggskriteriene: Godkjenning av bearbeidede mat- og fôrprodukter etter matloven, men forbud mot den samme GMO til utsetting etter genteknologiloven kan være aktuelt.
Ikke påvist helserisiko, men miljørisiko +/- negative konsekvenser med hensyn til tilleggskriteriene: Godkjenning av bearbeidede mat- og fôrprodukter etter matloven aktuelt, men forbud mot den samme GMO til utsetting etter genteknologiloven.
Påvist helserisiko +/- særnorsk miljørisiko og +/- negative konsekvenser med hensyn til tilleggskriteriene: Forbud mot bearbeidede mat- og fôrprodukter etter matloven, og forbud mot den samme GMO til utsetting etter genteknologiloven.
I tillegg kommer at vurderingskriteriene bærekraft, samfunnsnytte og etikk etter genteknologiloven kan innebære at en GMO godkjennes etter genteknologiloven, selv om det er en viss helse- og/eller miljørisiko, dersom et eller flere av tilleggskriteriene veier tungt nok og trekker i positiv retning.
Tabell 6.2 viser at det i Norge vil fattes to vedtak for samme GMO ved søknad etter nasjonalt regelverk: Bruksområde prosessert/bearbeidet mat og fôr etter matloven, og bruksområde utsetting etter genteknologiloven (import av levende GMO og evt. dyrking). Avhengig av om det påvises forhold i risikovurderingen og/eller konsekvenser med hensyn til etikk, bærekraft og samfunnsnytte for levende GMO, er flere vedtaksalternativer mulige. I tillegg kommer at vurderingskriteriene bærekraft, samfunnsnytte og etikk etter genteknologiloven kan innebære at en GMO godkjennes etter genteknologiloven selv om det er en viss risiko, dersom ett eller flere av tilleggskriteriene veier tungt nok og trekker i positiv retning.
Tabell 6.2 Søknad til Norge om godkjenning av alle bruksområder for en GMO til bruk som mat og fôr etter nasjonalt regelverk. Det kan fattes to vedtak for den samme GMO med hjemmel i hhv. matloven og genteknologiloven, som kan være like eller ulike.
Påvist helserisiko | Påvist miljørisiko | Konsekvens tilleggskriterier | Vedtak matloven | Vedtak gt-loven |
|---|---|---|---|---|
Nei | Nei | Ikke-negative | Godkjenning | Godkjenning |
Nei | Nei | Negative | Godkjenning | Forbud kan være aktuelt |
Nei | Ja | Negative/positive | Godkjenning kan være aktuelt | Forbud |
Ja | Ja/Nei | Negative/positive | Forbud | Forbud |
6.10.5 Vedtak etter dagens organisering av forvaltningen – søknader etter GM mat- og fôrforordningen
Dersom Norge får gjennomslag for utkast til tilpasningstekst, vil Norge etter en innlemming av GM-pakken antakelig ha rett til å fastsette nasjonale vedtak om godkjenning av, eller forbud mot den enkelte GMO hvis dette gjøres kort tid etter at EU har foretatt en beslutning. GMO til mat og fôr som godkjennes i EU etter GM mat- og fôrforordningen vil derfor ikke automatisk være godkjent i Norge.
Hvis dagens nasjonale forvaltning av genmodifisert mat og fôr opprettholdes også etter innlemming av GM-pakken, må norske myndigheter sikre at vedtakene under ulikt regelverk samordnes slik at de treffes samtidig og etter en enhetlig og helhetlig forståelse av GM mat- og fôrforordningen. Dette for å sikre mest mulig like konkurranseforhold for norske virksomheter. Én søknad til EU vil da behandles av to instanser og etter to regelverk i Norge, men det må sikres at det blir en enhetlig og helhetlig forvaltning selv om det fattes to vedtak for den samme GMO basert på ulike bruksområder. Basis vil være forordningene i GM-pakken og kravene til miljørisikovurderingen i utsettingsdirektivet, men med en vurdering av tilleggskriteriene for de levende produktene etter genteknologiloven.
Dersom foreliggende utkast til tilpasningstekst vedtas, vil utfallet av nasjonal søknadsbehandling etter en innlemming av GM-pakken i hovedsak bli lik som i dag. Eventuelle forbud må antakelig begrunnes med føre-var etter artiklene 53 og 54 i General Food Law, og Norge må da på sikt sende dokumentasjon som viser at det er grunn til å opprettholde slike forbud. Vurdering av eventuelle forbud med begrunnelse i bærekraft, samfunnsnytte og etikk, vil være som tidligere.
Tabell 6.3 viser at det også vil fattes to vedtak i Norge for samme GMO ved søknad til EU, hvis Norge gjennomfører GM-pakken både i matloven og genteknologiloven: Bruksområde prosessert/bearbeidet mat og fôr etter matloven og bruksområde utsetting etter genteknologiloven (import av levende GMO og evt. dyrking). Avhengig av om det påvises særnorske forhold i risikovurderingen og/eller konsekvenser med hensyn til etikk, bærekraft og samfunnsnytte for levende GMO, er flere vedtaksalternativer mulige. I tillegg kommer at tilleggskriteriene etter genteknologiloven kan innebære at en GMO godkjennes etter genteknologiloven selv om det er en viss risiko, dersom ett eller flere av de øvrige kriteriene veier tungt nok.
Tabell 6.3 Søknad til EU om godkjenning av alle bruksområder for en GMO til bruk som mat og fôr i en fremtidig situasjon der Norge har gjennomført GM-pakken både i matloven og genteknologiloven. Det kan fattes to vedtak for den samme GMO med hjemmel i hhv. matloven og genteknologiloven, som kan være like eller ulike.
Påvist særnorsk helserisiko | Påvist særnorsk miljørisiko | Konsekvens tilleggskriterier | Vedtak matloven | Vedtak genteknologiloven |
|---|---|---|---|---|
Nei | Nei | Ikke negative | Godkjenning | Godkjenning |
Nei | Nei | Negative | Godkjenning | Forbud kan være aktuelt |
Nei | Ja | Negative/positive | Godkjenning kan være aktuelt | Midl. forbud med hj. i føre-var (Food Law art. 53 og 54) |
Ja | Ja/Nei | Negative/positive | Forbud | Midl. forbud med hj. i føre-var (Food Law art. 53 og 54) |
6.10.6 Nærmere om konkrete koblinger
I Norge er det koblinger og overkrysninger på flere av forvaltningsområdene på området genmodifisert mat og fôr:
Regelverk for godkjenning av genmodifisert mat og fôr som ikke er levende (= død mat, for eksempel maismel, soyaolje etc.): Ansvarlig forvaltningsmyndighet er Mattilsynet. Regelverk under matloven relaterer seg til definisjonen av GMO i genteknologiloven. Dette er norsk regelverk som ligner på EUs regelverk, fordi EUs regelverk ennå ikke er innlemmet i EØS-avtalen. Det er ingen godkjente, genmodifiserte mat- eller fôrprodukter i Norge, men Mattilsynet behandler våren 2023 den første søknaden til Norge om godkjenning av en rapsolje fra GMO raps til bruk i fiskefôr.
Regelverk for godkjenning av levende GMO til mat og fôr: Ansvarlig myndighet er Klima- og miljødepartementet. Genteknologiloven forvaltes av Klima- og miljødepartementet (omsetning) og Miljødirektoratet (forsøksutsettinger og all annen bruk). Det er ikke søkt godkjenning for import eller dyrking/produksjon av levende GMO i Norge til mat og fôr. Miljødirektoratet har imidlertid 19. april 2023 mottatt en søknad om feltforsøk med genredigert laks. Genteknologiloven er en gjennomføring av EUs utsettingsdirektiv og har også noen nasjonale regler. Nasjonal godkjenning etter genteknologiloven vil nesten alltid være relevant for annet regelverk under matloven (merkekrav, sortsgodkjenning, importkontroll, plantehelse, dyrehelse, offentlig kontroll m.m.).
Regelverk for godkjenning av levende GMO som ikke er mat og fôr (for eksempel avskårne GMO-nellikblomster): Ansvarlig myndighet er Klima og miljødepartementet. Genteknologiloven anvendes og forvaltes av Klima- og miljødepartementet og Miljødirektoratet. Dette er EØS-regelverk, altså likt i EU og i EFTA-landene. For omsetningssøknader gjelder en felles prosedyre fastsatt i utsettingsdirektivet. Når en GMO blir godkjent etter denne prosedyren er den godkjent i hele EØS-området, også i Norge, med mindre Norge nedlegger et eksplisitt forbud. I dag er det lov å selge seks ulike typer GMO-nelliker som er genmodifisert for å få en lilla farge. Feltforsøk i forsøksøyemed med GMO (også GMO-er som kan inngå i matkjeden) godkjennes av miljømyndighetene i hvert enkelt land, i Norge av Miljødirektoratet. Koblingspunktet mot matloven vil være mht. importkontroll og plantehelseregelverk. All import av planter inkludert prydplanter skal meldes Mattilsynet i hht. regelverk om plantehelse og sjekkes opp mot skadegjørere, merkekrav mv. Både Miljødirektoratet og Mattilsynet fører tilsyn.
Regelverk for merking av godkjent mat og fôr: Ansvarlig forvaltningsmyndighet er Mattilsynet. Regler for merking av levende GMO mat og fôr og prosesserte produkter fra GMO, er hjemlet i matloven. Reglene er de samme som i EU, og gjelder godkjente produkter etter både matloven og genteknologiloven
Regelverk for sameksistenstiltak (dyrkingsregler): Ansvarlig forvaltningsmyndighet er Mattilsynet. Utkast til regler for å hindre innblanding av GMO i konvensjonelle og økologiske avlinger ved dyrking av godkjente GMO er utviklet av Mattilsynet og må forskriftsfestes hvis/når det blir aktuelt å dyrke GMO. Reglene vil gjelde uavhengig av om arten som dyrkes skal brukes til mat eller til annen bruk, og omhandler blant annet avstandskrav for ulike GMO til konvensjonelle og økologiske avlinger samt kompetansekrav til bønder.
Regelverk for grensekontroll av ikke-godkjent GMO til mat og fôr: Ansvarlig forvaltningsmyndighet er Mattilsynet. Regelverket gjelder både levende og prosessert GMO. Dette er EØS-regelverk, da hjemmelsgrunnlaget er kontrollforordningen som er gjennomført under matloven. I EU er det nullaksept for genmodifisert mat og fôr som ikke er godkjent etter unionsregelverket, også om de er helse- og miljørisikovurdert og godkjent i tredjeland. Her ligger føre-var-prinsippet implisitt til grunn, ved at tredjelands risikovurdering ikke oppfattes som tilstrekkelig og at man derfor »påberoper» en grad av vitenskapelig usikkerhet (man mangler kunnskap). Grensekontroll av ris og risprodukter fra Kina omfatter krav om analyse og myndighetssertifikater fra Kina samt analyse og myndighetsfrigivelse fra importlandet. Kravene er så strenge at import av slike produkter i praksis har blitt forbudt. Det har vært gjennomført flere slike kontrollmekanismer med basis i Food Law’s føre-var-prinsipp, blant annet på ulike GMO-maisvarianter fra USA, linfrø fra Canada og ulike risvarianter fra Kina.
Regelverk for sortsgodkjenning: Ansvarlig forvaltningsmyndighet er Mattilsynet. For å bli godkjent som ny sort, gjelder også GMO, må det være dokumentert at sorten er samfunnsnyttig i betydning av bedre enn tilsvarende sorter i minst én egenskap. Det skal også dokumenteres at sorten er genetisk stabil.
Landbruksrelatert miljørisiko knyttet til GMO: Matdepartementene og tidligere Miljøverndepartementet ble i 2006 enige om at matforvaltningen har et sektoransvar for agronomisk miljørisiko. Det er altså koblinger og overlapp i ansvaret for miljørisiko i matkjeden. Vitenskapskomiteen for mat og miljø (VKM) utfører miljørisikovurderinger av GMO etter oppdrag både fra Miljødirektoratet og Mattilsynet.
Helserisiko knyttet til levende og prosessert GMO: Mattilsynet er ansvarlig og koordinerende myndighet. Vurderingene i både genteknologi- og matregelverket er i hovedsak sammenfallende, og utføres av VKM etter oppdrag fra Mattilsynet.
Mulighet for nasjonalt dyrkingsforbud for GMO: Ansvarlig myndighet er Klima- og miljødepartementet. Medlemsland har i henhold til direktiv 2015/412 anledning til å legge ned nasjonale forbud mot dyrking av godkjent GMO basert på andre hensyn enn helse- og miljørisiko. Slike nasjonale forbud kan i EU nedlegges for godkjente GMO under GM mat- og fôrforordningen og utsettingsdirektivet. Listen over hensyn er ikke uttømmende og kan inkludere både etikk, samfunnsnytte og bærekraft og går således lenger enn de norske tilleggskriteriene. Dyrkings-/produksjonsdyrsøknader behandles etter genteknologiloven i Norge. Andre forhold knyttet til dyrking og begrensninger i dyrking, herunder hensyn som miljø, bærekraft og samfunnsnytte, er regulert av landbruks- og matforvaltningen.
Åpenhetsforordningen: Endrer seks rettsakter underliggende matloven samt utsettingsdirektivet. Endringen i utsettingsdirektivet gjør at også genteknologiloven måtte endres. Proposisjon med forslag om innlemmelse av åpenhetsforordningen i EØS-avtalen og endring av genteknologiloven er fremmet for Stortinget, som har gitt sitt samtykke og godkjent lovforslaget. I EU endres også GM mat- og fôrforordningen med åpenhetsforordningen.
Tabell 6.4 viser en oppsummering av koblingspunktene omtalt over.
I tillegg til ovenstående koblingspunkter, har noen GMO-er egenskaper som kan sammenliknes med produkter som er et resultat av mer tradisjonell landbruksteknologi. Ett eksempel er det såkalte Bt-proteinet, som kan uttrykkes i genmodifisert mais for å gi insektsresistens. Det aktuelle genet i disse maisplantene er hentet fra en jordbakterie, og denne Bt-bakterien kan alene brukes som et plantevernmiddel mot insekter (bakterien er selv ingen GMO). Både GMO-en og plantevernmiddelet må imidlertid risikovurderes, og begge er underlagt godkjenningsordninger i henholdsvis genteknologiregelverk og matregelverk. Også de genetiske endringene som er gjort i de enkleste genredigerte organismene kan sammenliknes med organismer som utvikles ved tradisjonell foredling. Selv om resultatorganismene da kan likne på hverandre, er veien fram til sluttresultatet forskjellig, og hvilket regelverk som kommer til anvendelse avhenger derfor av teknologien som er brukt.
Tabell 6.4 Oversikt over de viktigste koblingspunktene mellom matloven og genteknologiloven.
Prosessert GMO mat og fôr | Levende GMO mat og fôr | Levende GMO annet enn mat og fôr | |
|---|---|---|---|
GMO-definisjon | genteknologiloven | genteknologiloven | genteknologiloven |
Godkjenningskrav | matloven | genteknologiloven | genteknologiloven |
BSE | - | genteknologiloven | genteknologiloven |
Koordinering helserisiko | matloven | matloven | genteknologiloven |
Koordinering miljørisiko | - | genteknologiloven | genteknologiloven |
Koordinering agronomisk miljørisiko | matloven | matloven | matloven |
Merkekrav | matloven | matloven | genteknologiloven |
Sortsgodkjenning | - | matloven | matloven |
Merkekrav såvarer | - | matloven | matloven |
Nasjonalt dyrkingsforbud | - | genteknologiloven | genteknologiloven |
Nasjonale dyrkingsregler | matloven | matloven | matloven |
Offentlig kontroll | matloven | matloven | genteknologiloven |
Grensekontroll | matloven | matloven | matloven |
Et annet eksempel er organismer som har fått tilført transiente (forbigående) genetiske egenskaper ved hjelp av for eksempel RNAi-molekyler. RNAi kan da virke nærmest som et sprøytemiddel, hvis det brukes på planter i en åker. I fall man ikke lander på at en slik organisme skal reguleres som en GMO, behøver ikke «situasjonen» som sådan være uregulert. Alternative reguleringsmåter under annet regelverk kan være aktuelt, slik som godkjenningsordningen for plantevernmidler.
Fotnoter
Se kapittel 12.2.2 for nærmere omtale.
Se mer informasjon om dette i kapittel 5, punkt 5.6.1.
https://www.fao.org/fao-who-codexalimentarius/en/, se også nærmere omtale i kapittel 5.4.1.
International Union for the Protection of New Varieties of Plants (UPOV).
https://www.regjeringen.no/contentassets/38f9ecfe9a524c0e9fee99011d7e41f3/20170705---gmo-saksbehandlingsrutiner.pdf
CLYNAV; INN: deoxyribonucleic acid (europa.eu)
EFSA Science, safe food, sustainability (europa.eu).
Microsoft Word – Bærekrafthefte revidert BN 06 UTEN VIS ENDRINGER.doc (bioteknologiradet.no))
Se nærmere omtale i kapittel 5.6.1.
EUROPAPARLAMENTS- OG RÅDSDIREKTIV (EU) 2015/412 av 11. mars 2015 om endring av direktiv 2001/18/EF med hensyn til medlemsstatenes mulighet til å begrense eller forby dyrking av genmodifiserte organismer (GMO) på sitt territorium (*)
Frivillig skjema utarbeidet i interplay-samarbeidet: MEDICINAL PRODUCTS FOR HUMAN USE CONTAINING OR CONSISTING OF GMOS: INTERPLAY BETWEEN THE EU LEGISLATION ON MEDICINAL PRODUCTS AND GMOS
Europaparlaments- og rådsforordning (EF) nr. 178/2002 av 28. januar 2002 om fastsettelse av allmenne prinsipper og krav i næringsmiddelregelverket, om opprettelse av Den europeiske myndighet for næringsmiddeltrygghet og om fastsettelse av framgangsmåter i forbindelse med næringsmiddeltrygghet.
https://lovdata.no/dokument/SF/forskrift/2008-12-22-1620.
https://www.mattilsynet.no/om_mattilsynet/import_og_eksport/rasff/, RASFF Mattilsynet
https://www.wto.org/english/res_e/booksp_e/dispu_settl_1995_2017_e.pdf, WTO Dispute Settlement: One-Page Case Summaries – 1995–2016
Se konsolidert versjon av forordningen på engelsk her: CL2003R1829EN0030010.0001_cp 1.1 (europa.eu)
Se konsolidert versjon på engelsk her: CL2003R1830EN0020010.0001_cp 1.1 (europa.eu)
Commission Regulation (EC) No 65/2004, Commission Regulation (EC) No 641/2004, Commission Recommendation 2004/787/EC, Commission Regulation (EC) No 1981/2006, Regulation (EC) No 298/2008, Regulation (EC) No 1137/2008, Commission Regulation (EU) No 619/2011, Commission Implementing Regulation (EU) No 503/2013, Commission Implementing Regulation (EU) No 120/2014, Regulation (EU) 2019/1381.
Se norsk oversettelse her: 32001l0018.pdf (lovdata.no), konsolidert versjon på engelsk her: CL2001L0018EN0070010.0001.3bi_cp 1.1 (europa.eu).
Kommisjonsforordning (EF) nr. 65/2004 av 14. januar 2004 om etablering av et system for utvikling og tildeling av unike koder for genmodifiserte organismer.
Kommisjonsrekommandasjon 2010/722/EU av 13. juli 2010 om retningslinjer for utvikling av nasjonale sameksistenstiltak for å unngå utilsiktet innblanding av GMO i konvensjonelle og økologiske avlinger.
Europaparlaments- og rådsforordning (EU) nr. 2017/625 av 15. mars 2017 om offentlig kontroll og annen offentlig virksomhet som gjennomføres for å sikre anvendelsen av næringsmiddel- og fôrvareregelverket samt regler for dyrs helse og velferd, plantehelse og plantevernmidler, om endring av europaparlaments- og rådsforordning (EF) nr. 999/2001, (EF) nr. 396/2005, (EF) nr. 1069/2009, (EF) nr. 1107/2009, (EU) nr. 1151/2012, (EU) nr. 652/2014, (EU) 2016/429 og (EU) 2016/2031, rådsforordning (EF) nr. 1/2005 og (EF) nr. 1099/2009 samt rådsdirektiv 98/58/EF, 1999/74/EF, 2007/43/EF, 2008/119/EF og 2008/120/EF og om oppheving av europaparlaments- og rådsforordning (EF) nr. 854/2004 og (EF) nr. 882/2004, rådsdirektiv 89/608/EØF, 89/662/EØF, 90/425/EØF, 91/496/EØF, 96/23/EF, 96/93/EF og 97/78/EF og rådsbeslutning 92/438/EØF.
https://www.regjeringen.no/no/dokumenter/departementenes-eos-arbeid/id2864387/, Departementenes EØS-arbeid (regjeringen.no).
Høring: Risikovurdering av Aquaterra® olje fra genmodifisert raps Mattilsynet
Ot.prp. nr. 100 (2002–2003), s 142.
Forskrift 28. november 2014 om matinformasjon til forbrukerne (matinformasjonsforskriften).
Forskrift 2. april 2011 nr. 360 om merking og omsetning av fôrvarer.
Forskrift 13. september 1999 om såvarer.
Forskrift 4. oktober 2005 nr. 1103 om økologisk produksjon og merking av økologiske landbruksprodukter og næringsmidler.
Forskrift 1.oktober 1999 nr. 1069 om prøving og godkjenning av plantesorter.
Forskrift 1. desember 2000 om planter og tiltak mot planteskadegjørere.
Forskrift 12. januar 2012 nr. 35 om særskilte beskyttelsestiltak ved import av ris og risprodukter fra Kina.
Forskrift 22. desember 2008 nr. 1620 om allmenne prinsipper og krav i næringsmiddelregelverket (matlovsforskriften); Forskrift 12. april 2005 nr. 319 om tilsetningsstoffer til bruk i fôrvarer; Forskrift 6. juni 2011 nr. 669 om aroma og næringsmiddelingredienser med aromagivende egenskaper til anvendelse i og på næringsmidler; Forskrift 21. desember 1993 nr. 1381 om materialer og gjenstander i kontakt med næringsmidler; Forskrift 6.juni 2011 nr. 666 Forskrift om godkjenning av enzymer mm. i næringsmidler; Forskrift 6. mai 2015 nr. 455 om plantevernmidler; Forskrift 25. juli 2017 nr. 1215 om ny mat.
https://gmo-crl.jrc.ec.europa.eu/gmomethods/
JRC116289-GE-report-ENGL.pdf (europa.eu)
Se omtale av forskningsprogrammet her: Horizon Europe Framework programme – deteksjon av organismer og produkter fra nye genomteknikker
Se mer om prosjektet på Forskningsrådets hjemmesider: https://prosjektbanken.forskningsradet.no/project/FORISS/301911.
European Network of GMO Laboratories (ENGL). Definition of minimum performance requirements for analytical methods of GMO testing. 20 October 2015 (JRC95544). https://gmo-crl.jrc.ec.europa.eu/doc/MPR%20Report%20Application%2020_10_2015.pdf, MPR Report Application 20_10_2015.pdf (europa.eu)
https://inspirasjon.interflora.no/blomsterpleie/sett-farge-pa-maten-med-spiselige-blomster. Sett farge på maten med spiselige blomster Interflora Norge.
https://norecopa.no/media/6332/endelig-instruks-for-forsoksdyrforvaltningen.pdf
https://www.mattilsynet.no/dyr/forsoksdyr/soke-godkjenning-som-forsoksdyrvirksomhet?kapittel=1-slik-sker-du-om-godkjenning-eller-endring-av-virksomhet. Forsøksdyr-forvaltningens tilsyns- og søknadssystem.
https://www.forskningsetikk.no/retningslinjer/nat-tek/etiske-retningslinjer-for-bruk-av-dyr-i-forskning/
Tekst delvis hentet fra Bruk av dyr i forsøk i 2020 (mattilsynet.no)
Mattilsynets arbeid med dyrevelferd 2022 (enonic.cloud)
bruk-av-dyr-i-forsoek-2022.pdf (norecopa.no)
Arsrapport-2020-ENDELIG.pdf (forsoksdyrkomiteen.no)
DS291/37, se WTO Dispute Settlement: One-Page Case Summaries – 1995–2016.
Risk assessment of Aquaterra® oil for its intended use as ingredient in fish feed. VKM Report 2023: 8.
Kommisjonens gjennomføringsforordning (EU) nr. 503/2013 av 3. april 2013 om søknader om godkjenning av genetisk modifiserte næringsmidler og fôrvarer i henhold til europaparlaments- og rådsforordning (EF) nr. 1829/2003 og om endring av kommisjonsforordning (EF) nr. 641/2004 og (EF) nr. 1981/2006.